Einleitung
Gewebe, Zellen und die kleineren Strukturen in Zellen (Organellen) sind meist Wasser und daher transparent. Imaging winzige durchsichtige Beutel mit Wasser führt zu Bildern, die nicht viele Informationen enthalten, und in der Mikroskopie ist es wichtig, eine Art Kontrast oder Fleck zu haben, die Bereiche der Probe Farbe geben und sie viel leichter zu sehen machen. Was ist außerdem, wenn Sie nur einige der kleineren Strukturen in einer Zelle wie einen Zellkern oder eine Zellmembran abbilden möchten? Das Einfärben der gesamten Zelle würde es unmöglich machen, die Bereiche zu lokalisieren, an denen Sie interessiert sind.
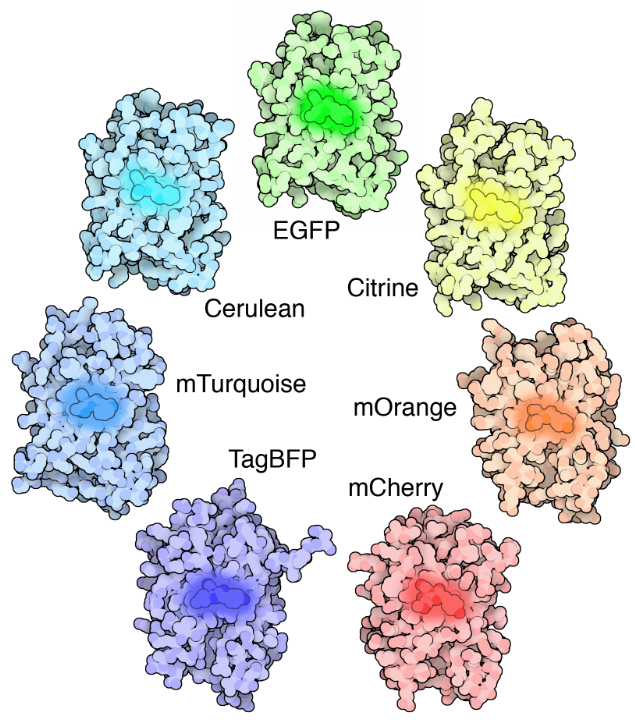
Fluoreszenz löst diese beiden Probleme von Kontrast und Lokalisierung. Fluoreszenz ist, wo ein Objekt Licht emittiert, nachdem es Licht absorbiert hat. Viele verschiedene Objekte zeigen Fluoreszenz, wie Mineralien (das Wort Fluoreszenz kommt vom Mineral Fluorit), Tiefseefische (am bekanntesten die Qualle Aequorea victoria, aus der grün fluoreszierendes Protein (GFP) entdeckt wurde), Pflanzen, Chemikalien und vieles mehr.
Fluoreszierende Moleküle (bekannt als Fluorophore) werden zur Markierung von Proben verwendet, und Fluorophore sind verfügbar, die Licht in praktisch jeder Farbe emittieren. In einem Fluoreszenzmikroskop wird eine Probe mit einem Fluorophor markiert, und dann wird ein helles Licht (Anregungslicht) verwendet, um die Probe zu beleuchten, die Fluoreszenz (Emissionslicht) abgibt. Auf diese Weise werden Proben stark zum schwarzen Hintergrund kontrastiert, da der Fluorophor ein helles Licht emittiert. Durch die Lokalisierung dieser Fluorophore im interessierenden Bereich kann ein klares Bild eines beliebigen Teils einer Zelle aufgenommen werden, was die Fluoreszenzmikroskopie zu einem leistungsstarken Werkzeug für die Biowissenschaften macht.
Brightfield vs Fluorescence Imaging
Bei der Hellfeldmikroskopie wird die Probe mit weißem transmittiertem Licht beleuchtet. Dies erzeugt eine gleichmäßige Ausleuchtung der Probe unter dem Mikroskop, um kontrastreiche, gefärbte oder natürlich pigmentierte Proben zu beobachten. Das Hellfeld reicht jedoch nicht aus, um zwischen transparenten / transluzenten, ungefärbten Zellen oder zellulären Strukturen zu unterscheiden, um Prozesse von Interesse zu untersuchen.
Die Fluoreszenzmikroskopie beruht auf der Verwendung von Fluorophoren, Molekülen, die Licht einer bestimmten sichtbaren Wellenlänge emittieren, wenn sie Licht einer anderen Wellenlänge ausgesetzt werden. Wenn diese Fluorophore an eine gezielte Struktur von Interesse gebunden sind, können Photonen, die von dem Fluorophor emittiert werden, verwendet werden, um diese Struktur von Interesse zu visualisieren. Der Vorteil der Fluoreszenzmikroskopie besteht darin, dass die Zielstrukturen beleuchtet werden, während die unerwünschten Bereiche der Probe wenig bis gar keine Fluoreszenz aufweisen, was ein einfaches Targeting und Imaging ermöglicht.
Warum Moleküle fluoreszieren
Der Ursprung der Fluoreszenz sind Elektronen, die sich frei um den aktiven Fluorophor bewegen und absorbierte Energie freisetzen, wie in Abb.2.
Vor der Anregung befinden sich Elektronen im niedrigsten Energiezustand, der ihnen zur Verfügung steht – dem Grundzustand (S0). Wenn ein Elektron mit einem Photon eines bestimmten Energiebereichs getroffen wird, absorbiert das Elektron die Energie des Photons und springt in einen höheren Energiezustand (S1, S2 oder S3). Um in den Grundzustand (S0) zurückzukehren, gibt das Elektron die zusätzliche Energie als Emission eines Photons frei. Die Energie dieses Photons ist geringer als die Anregungsenergie, so dass es eine längere Wellenlänge hat. Aus diesem Grund hat Emissionslicht eine längere Wellenlänge als das Anregungslicht und kann als eine andere Farbe erscheinen.
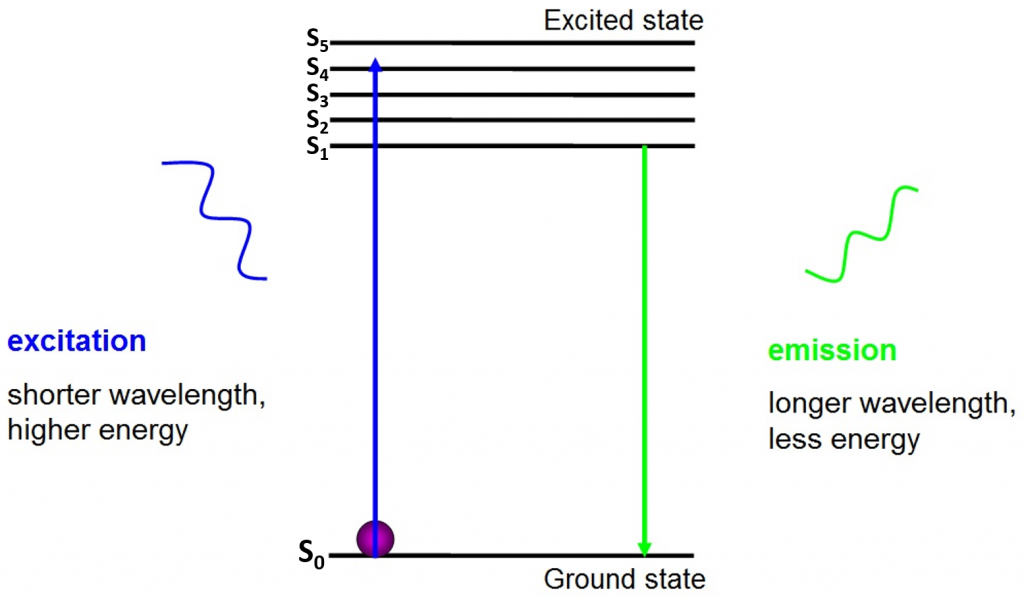
Das emittierte Photon befindet sich normalerweise im sichtbaren Spektrum und kann unter einem Mikroskop betrachtet werden, wenn genügend angeregte Fluorophore vorhanden sind. Die Wellenlänge des freigesetzten Photons ist für jeden Fluorophor spezifisch und diese Vorhersagbarkeit ermöglicht eine einfache Fluoreszenzbildgebung.
Fluoreszenzintensitätsfaktoren
Während Fluorophore Fluoreszenz einer vorhersagbaren Wellenlänge emittieren können, ist es auch wichtig zu wissen, welche Faktoren die Fluoreszenzintensität steuern. Ohne eine ausreichend intensive Lichtemission ist die Fluoreszenz mit einem Mikroskop nicht nachweisbar.
Quantenausbeute
Die Quantenausbeute (ϕ) eines Fluorophors ist das Verhältnis der Anzahl der freigesetzten Photonen zur Anzahl der absorbierten Photonen. Die Quantenausbeute wird oft als Wert von 0-1 ausgedrückt, wobei 1 100% Effizienz der Photonenkonversion ist. Es ist auch wichtig zu beachten, dass jeder Fluorophor einen einzigartigen pH-Wert, Ionenstärke und Temperatur für eine optimale Fluoreszenzeffizienz hat.
Extinktionskoeffizient
Jeder Fluorophor hat eine andere Fähigkeit, Photonen zu absorbieren, selbst wenn sie sich in einem geeigneten Wellenlängenbereich befinden, um ihn anzuregen. Wenn ein Fluorophor einem Photon ausgesetzt wird, das seiner Anregungswellenlänge angemessen entspricht, ist die Wahrscheinlichkeit, dass ein Photon absorbiert wird, eine messbare Eigenschaft und bekannt als Extinktionskoeffizient (ε).
Die Quantenausbeute und der Extinktionskoeffizient eines Fluorophors werden oft zusammen angezeigt, um zu beschreiben, wie hell der Fluorophor in experimentellen Umgebungen ist.
Fluoreszenzlebensdauer
Wenn ein Fluorophorelektron ein Photon absorbiert, setzt es nicht sofort ein längerwelliges Photon frei. Es ist bekannt, dass die Freisetzung von Energie zwischen angeregten Energiezuständen unterschiedlich lange dauert. Die Zeit, die ein Elektron im angeregten Zustand verbringt, bevor es ein Photon freisetzt und in den Grundzustand zurückkehrt, ist ein Maß für seine Fluoreszenzlebensdauer. Die Lebensdauer jedes Fluorophors ist einzigartig und kann experimentell gemessen werden. Bei der experimentellen Verwendung von Fluoreszenzfarbstoffen ist es hilfreich, ihre Lebensdauer zu verlängern, insbesondere für Anwendungen, die eine hohe Geschwindigkeit erfordern, wie z. B. Calcium-Imaging-Neuronen.
Anregungswellenlängenintensität
Die meisten Fluoreszenzmikroskopie-Aufbauten enthalten eine Lichtquelle, die auf den gewünschten Wellenlängenbereich abgestimmt werden kann. Viele Leuchtstofflichtquellen können auch auf Anregungsintensität eingestellt werden, um die Anzahl der Photonen zu erhöhen, die sich durch den Lichtweg bewegen. In einer fluoreszenzmarkierten Probe, die ihrer Anregungswellenlänge ausgesetzt ist, wird nicht jeder Fluorophor gleichzeitig aktiviert. Durch Erhöhen der Anregungsintensität und Erhöhen der Anzahl der Photonen, die die Probe erreichen, besteht eine höhere Wahrscheinlichkeit, dass mehr Fluorophore angeregt werden.
Photostabilität
Photostabilität ist die Fähigkeit eines Moleküls oder Organismus, Schäden zu widerstehen. In der Fluoreszenzmikroskopie hören Fluorophore schließlich auf, entgegenkommende Photonen zu absorbieren und gehen in einen dauerhaften dunklen Zustand über. Wenn ein Organismus im dunklen Zustand mehr Fluorophore sammelt, verringert sich das Erscheinungsbild des markierten Ziels, und die Probe soll photobleichen. In der Fluoreszenzmikroskopie werden häufig Schritte unternommen, um die Menge an Photobleichen zu reduzieren, die während des Experimentierens auftritt. Einige Maßnahmen umfassen eine Verringerung der Lichtintensität, die mit der Probe interagiert, und die Verwendung spezieller Fluoreszenzfarbstoffe, die nicht so lange aktiv bleiben wie andere Farbstoffe.
Fluoreszenzmikroskopie
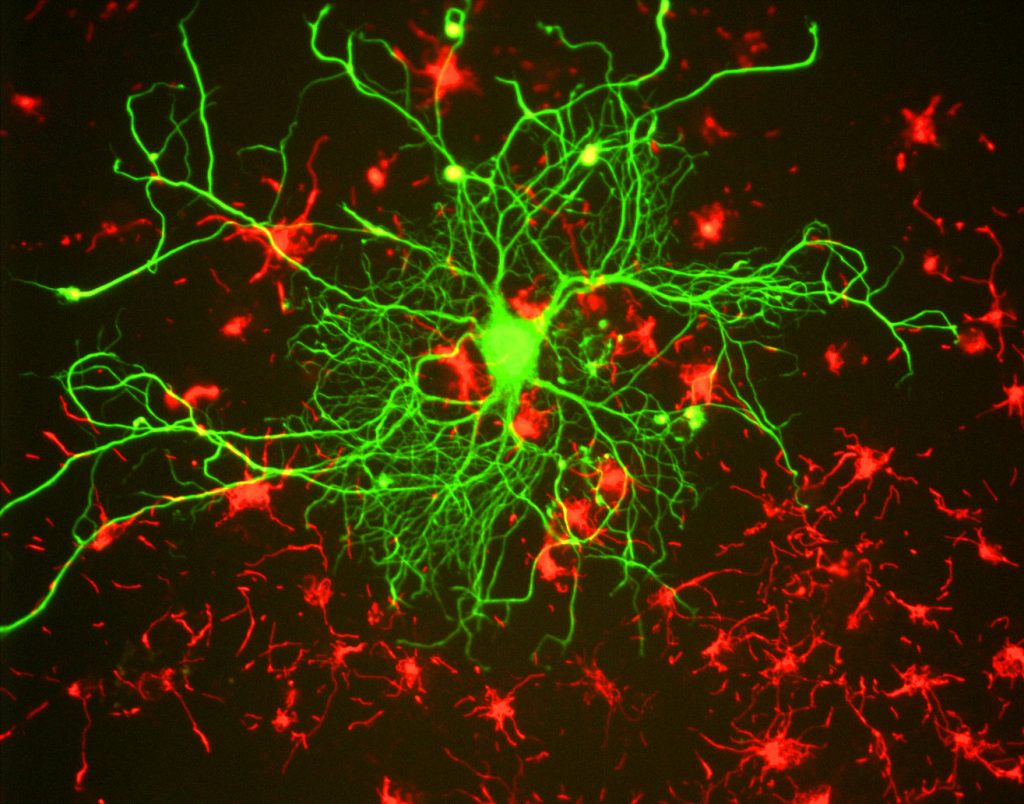
Für Forscher ist der Hauptvorteil der Fluoreszenz die Möglichkeit, Fluoreszenzmikroskopie zu verwenden, bei der Proben mit einer fluoreszierenden Substanz wie einem Farbstoff, Antikörper oder Protein markiert / gefärbt werden, wodurch Bilder kontrastiert werden können. Durch die Ausrichtung auf diese fluoreszierenden Etiketten können Forscher auswählen, was sie sehen möchten. Dies ist in Fig.3, wo ein Neuron deutlich unter Astrozyten zu sehen ist, da sie mit verschiedenen Farben von Fluoreszenzmarkern markiert sind.
Im Allgemeinen wird für die Fluoreszenzmikroskopie eine Probe mit Fluoreszenzmarkern markiert (typischerweise spezifisch für bestimmte Teile der Probe). Die Probe wird dann mit der spezifischen Anregungswellenlänge für den Fluorophor beleuchtet, und die resultierende Emissionsfluoreszenz wird vom Detektor, normalerweise einer empfindlichen wissenschaftlichen Kamera, empfangen.
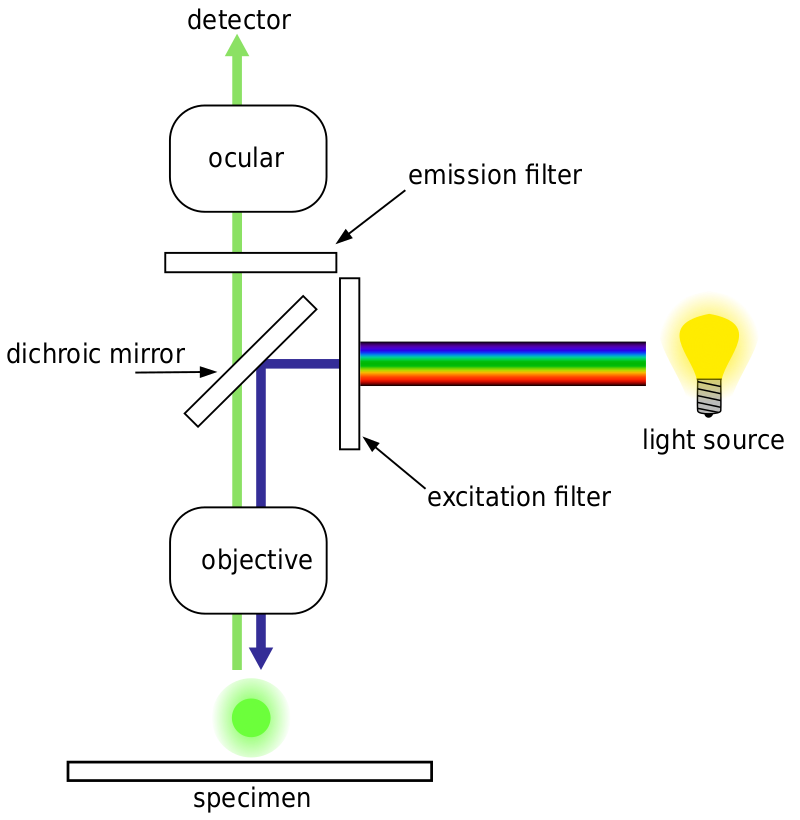
Die meisten Fluoreszenzmikroskope sind Epifluoreszenzmikroskope, bei denen Anregung und Emission über denselben Lichtweg erfolgen. Sowohl die Anregungsbeleuchtung als auch die emittierte Fluoreszenz passieren das Mikroskopobjektiv und werden üblicherweise gefiltert, um nur die Fluoreszenz zu detektieren. Dieser Aufbau ist in Fig.4.
Autofluoreszenz
Einige Strukturen, biologische Organismen und allgemeine Mikroskopieproben können auf natürliche Weise Fluoreszenz aufweisen, die als Autofluoreszenz bezeichnet wird. Dies unterscheidet sich von Fluoreszenz von markierten Proben, aber es teilt oft ähnliche Wellenlängen, was bedeutet, dass Autofluoreszenz-Mikroskopie-Proben künstlich hinzugefügt Fluoreszenz verdecken können und stört die Erkennung, Signal zu reduzieren. Es ist wichtig zu wissen, ob Ihre Proben Autofluoreszenz aufweisen, da dies die Fluoreszenzbildgebung beeinträchtigt, es sei denn, es werden bestimmte Wellenlängen verwendet, um dies zu vermeiden.
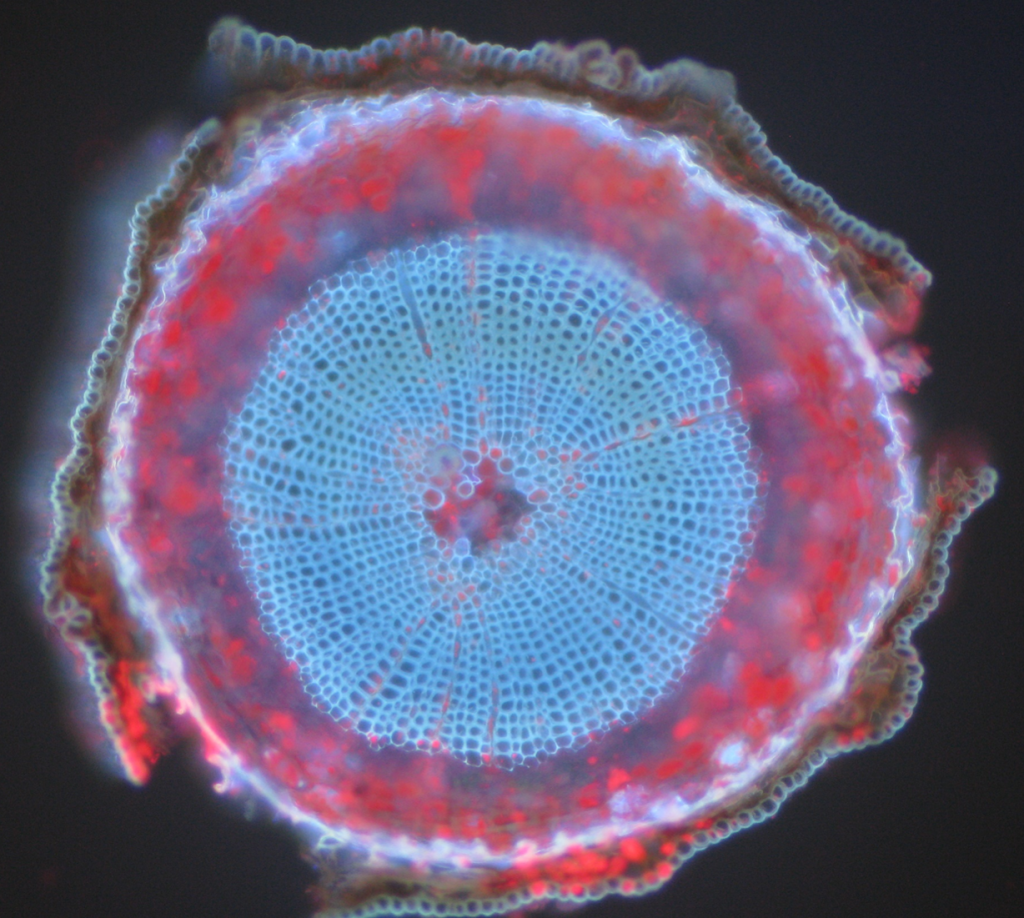
Häufige Beispiele für autofluoreszierende Objekte sind Mitochondrien, Lysosomen, Kollagen und einige Aminosäuren wie Tryptophan, Tyrosin und Phenylalanin. Vor allem Autofluoreszenz ist in Pflanzen aufgrund ihrer Verwendung von Chlorophyll und anderen fluoreszierenden Molekülen wie Ligninen und Carotinen üblich. Abb.5 zeigt die verschiedenen Farben der Autofluoreszenz aus einer unbeschrifteten Föhrenprobe.
Summary
Seit der Einführung des ersten Fluoreszenzfarbstoffs ist die Fluoreszenzmikroskopie ein häufig eingesetztes Werkzeug zur Visualisierung von Zellen und Zellstrukturen mit höherer Spezifität als herkömmliche Hellfeldmikroskopietechniken. Forscher canmanipulate die Struktur, die optischen Eigenschaften und die Sonde der Interessefluoreszenzexperimente, zum von relevanten Daten zu erhalten. Diese Flexibilität hat es Ermöglichtfluoreszenzmikroskopie, in viele Life-Science-Experimente einbezogen zu werden.
Je nach Art der Probe und des Fluorophors sollte eine wissenschaftliche Kamera sorgfältig ausgewählt werden, um die besten Abbildungsergebnisse zu erzielen.
- Lichtmikroskopie. (2009) Natur Meilensteine. MacMillan Publishers Limited, 6-22.
- Lavis, LD, & Raines, RT (2008). Helle Ideen für die chemische Biologie. ACS Chemische Biologie, 3 (3): 142-155.
- Liu, Y., Lilly, D. (2017) Kristallstrukturen, wenn Cyaninfluorophore auf das Ende doppelsträngiger RNA stapeln. Biophysical Journal, 113, (11): 2236-2343.
- Beresin, M. Y., Achilefu, S. (2010). Fluoreszenzlebensdauermessungen und biologische Bildgebung. Chemische Bewertungen, 110 (5): 2641-2684.
- Stockert, J., Blazquez-Castro, A. (2017) Fluoreszenzmikroskopie in den Lebenswissenschaften. Sharjah, Vereinigte Arabische Emirate. Bentham Science Publishers.
- Beresin, M. Y., & Achilefu, S. (2010). Fluoreszenzlebensdauermessungen und biologische Bildgebung. Chemische Bewertungen, 110 (5): 2641-2684.
- Denk, W., Strickler, J., Webb, W. (1990). Zwei-Photonen-Laser-Scanning-Fluoreszenzmikroskopie. Wissenschaft. 248, (1951): 73-76.
- So, P. (2002). Zwei-Photonen-Fluoreszenz-Lichtmikroskopie. In: Macmillan Publishing Group.
- Schermelleh, L., Heinztmann, R. und Leonardt, H. (2010). Ein Leitfaden zur superauflösenden Fluoreszenzmikroskopie. Das Journal für Zellbiologie 190 (2): 165-175.
- Betzig, E., Patterson, G.H., Sougrat, R., Lindwasser, O. W., Olenych, S., Bonifacino, J. S., Davidson, M. W., LippincottSchwartz, J., Hess, H.F. (2006) Darstellung von intrazellulären Leuchtstoffproteinen an der Nanometerauflösung. Wissenschaft. 313(5793): 1642-5
- Rost, MJ, Bates, M. & Zhuang, X. (2006) Subbeugungsgrenzenbildgebung durch stochastische optische Rekonstruktionsmikroskopie (STORM). Nat-Methoden. 2, (10):793-5.
- Rego, E., Shao, L., Macklin, J. Winoto, L., Johansson, G., Kamps-Hughes, N., Davidson, M. und Gustasson, M. (2010) PNAS. 109 Absatz 3: e135-a143.
- Jungmann, R., Avendaño, M. S., Woehrstein, J. B., Dai, M., Shih, W. M. & Yin, P. (2014) Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat-Methoden. 11(3): 313-318
- Jiang, X., Wang, L., Carroll, S., Chen, J., Wang, M., und Wang, J. (2018) Challenges and Opportunities for Small Molecule Fluorescent Probes in Redox Biology Applications. Antioxidants & Redox Signaling. Mary Ann Liebert, Inc.