Introduction
Les tissus, les cellules et les structures plus petites à l’intérieur des cellules (organites) sont principalement de l’eau et sont donc transparents. L’imagerie de petits sacs d’eau transparents donne des images qui ne contiennent pas beaucoup d’informations, et en microscopie, il est essentiel d’avoir une sorte de contraste ou de tache qui donnera des zones de couleur à l’échantillon et les rendra beaucoup plus faciles à voir. De plus, que se passe-t-il si vous voulez seulement imager certaines des plus petites structures à l’intérieur d’une cellule, comme un noyau ou une membrane cellulaire? La coloration de la cellule entière rendrait impossible la localisation des zones qui vous intéressent.
La fluorescence résout à la fois ces problèmes de contraste et de localisation. La fluorescence est l’endroit où un objet émettra de la lumière après avoir absorbé la lumière. De nombreux objets présentent une fluorescence, tels que des minéraux (le mot fluorescence provenant de la fluorite minérale), des poissons d’eau profonde (la plus célèbre est la méduse Aequorea victoria, à partir de laquelle la protéine fluorescente verte (GFP) a été découverte), des plantes, des produits chimiques et bien d’autres.
Des molécules fluorescentes (appelées fluorophores) sont utilisées pour marquer les échantillons, et des fluorophores sont disponibles qui émettent de la lumière dans pratiquement toutes les couleurs. Dans un microscope fluorescent, un échantillon est marqué avec un fluorophore, puis une lumière vive (lumière d’excitation) est utilisée pour éclairer l’échantillon, ce qui dégage une fluorescence (lumière d’émission). De cette manière, les échantillons sont très contrastés avec le fond noir car le fluorophore émet une lumière de couleur vive. En localisant ces fluorophores dans la zone d’intérêt, une image claire de n’importe quelle partie d’une cellule peut être prise, faisant de la microscopie à fluorescence un outil puissant pour les sciences de la vie.
Imagerie à fond clair vs Fluorescence
En microscopie à fond clair, l’échantillon est éclairé par une lumière blanche transmise. Cela crée un éclairage uniforme de l’échantillon au microscope pour observer des échantillons très contrastés, colorés ou naturellement pigmentés. Cependant, le fond clair n’est pas suffisant pour faire la distinction entre les cellules ou les structures cellulaires transparentes / translucides non colorées pour étudier les processus d’intérêt.
La microscopie à fluorescence repose sur l’utilisation de fluorophores, des molécules qui émettent une lumière d’une longueur d’onde visible spécifique lorsqu’elles sont exposées à une lumière d’une longueur d’onde différente. Lorsque ces fluorophores sont liés à une structure d’intérêt ciblée, les photons émis par le fluorophore peuvent être utilisés pour visualiser cette structure d’intérêt. L’avantage de la microscopie à fluorescence est que les structures ciblées sont éclairées tandis que les zones indésirables de l’échantillon ont peu ou pas de fluorescence, ce qui permet un ciblage et une imagerie faciles.
Pourquoi les molécules fluorent
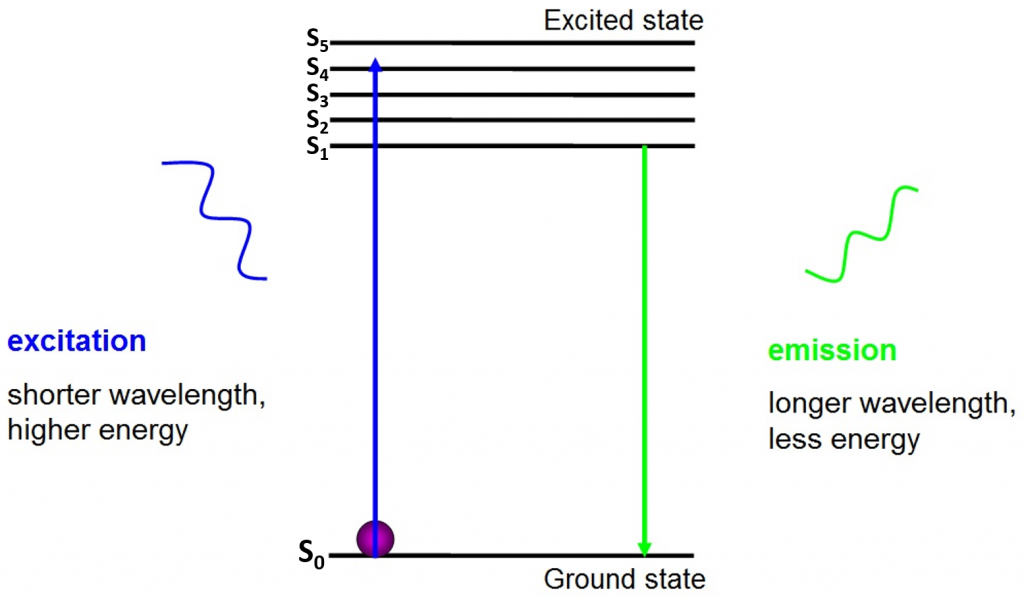
L’origine de la fluorescence est que les électrons se déplacent librement autour du fluorophore actif et libèrent de l’énergie absorbée, comme le montre la Fig.2.
Avant l’excitation, les électrons sont dans l’état d’énergie le plus bas à leur disposition – l’état fondamental (S0). Lorsqu’un électron est frappé par un photon d’une certaine plage d’énergie, l’électron absorbe l’énergie du photon et saute jusqu’à un état d’énergie plus élevé (S1, S2 ou S3). Pour revenir à l’état fondamental (S0), l’électron libère l’énergie supplémentaire sous forme d’émission d’un photon. L’énergie de ce photon est inférieure à l’énergie d’excitation, il a donc une longueur d’onde plus longue. C’est pourquoi la lumière d’émission a une longueur d’onde plus longue que la lumière d’excitation et peut apparaître sous une couleur différente.
Le photon émis est généralement dans le spectre visible et peut être vu au microscope s’il y a suffisamment de fluorophores excités. La longueur d’onde du photon libéré est spécifique à chaque fluorophore et cette prévisibilité permet une imagerie de fluorescence facile.
Facteurs d’intensité de fluorescence
Bien que les fluorophores puissent émettre une fluorescence d’une longueur d’onde prévisible, il est également important de savoir quels facteurs contrôlent l’intensité de fluorescence. Sans une émission de lumière suffisamment intense, la fluorescence ne sera pas détectable au microscope.
Rendement quantique
Le rendement quantique (ϕ) d’un fluorophore est le rapport entre le nombre de photons libérés et le nombre de photons absorbés. Le rendement quantique est souvent exprimé sous la forme d’une valeur comprise entre 0 et 1, 1 étant 100% d’efficacité de conversion de photons. Il est également important de noter que chaque fluorophore a un pH, une force ionique et une température uniques pour une efficacité de fluorescence optimale.
Coefficient d’extinction
Chaque fluorophore a une capacité différente à absorber les photons même s’ils se trouvent dans une plage de longueurs d’onde appropriée pour l’exciter. Si un fluorophore est exposé à un photon correspondant de manière appropriée à sa longueur d’onde d’excitation, la probabilité qu’un photon soit absorbé est une caractéristique mesurable et connue sous le nom de coefficient d’extinction (ε).
Le rendement quantique et le coefficient d’extinction d’un fluorophore sont souvent affichés ensemble pour décrire la luminosité du fluorophore dans des contextes expérimentaux.
Durée de vie de fluorescence
Lorsqu’un électron fluorophore absorbe un photon, il ne libère pas immédiatement un photon de longueur d’onde plus longue. La libération d’une certaine énergie entre les états d’énergie excités est connue pour prendre différentes longueurs de temps. Le temps qu’un électron passe à l’état excité avant de libérer un photon et de revenir à l’état fondamental est une mesure de sa durée de vie de fluorescence. La durée de vie de chaque fluorophore est unique et peut être mesurée expérimentalement. Lors de l’utilisation expérimentale de colorants fluorescents, leur durée de vie est utile, en particulier pour les applications nécessitant une vitesse élevée, telles que les neurones d’imagerie calcique.
Intensité de longueur d’onde d’excitation
La plupart des configurations de microscopie fluorescente comprennent une source de lumière qui peut être réglée pour produire la gamme de longueurs d’onde souhaitée. De nombreuses sources de lumière fluorescente peuvent également être ajustées pour l’intensité d’excitation afin d’augmenter le nombre de photons se déplaçant dans le trajet de la lumière. Dans un échantillon marqué par fluorescence qui est exposé à sa longueur d’onde d’excitation, chaque fluorophore ne sera pas activé en même temps. En augmentant l’intensité d’excitation et en augmentant le nombre de photons atteignant l’échantillon, il y a une plus grande probabilité que plus de fluorophores soient excités.
Photostabilité
La photostabilité est la capacité d’une molécule ou d’un organisme à résister aux dommages. En microscopie à fluorescence, les fluorophores finiront par cesser d’absorber les photons venant en sens inverse et passeront dans un état d’obscurité permanent. Comme un organisme rassemble plus de fluorophores à l’état sombre, l’apparence de la cible marquée diminue et l’échantillon est dit photoblanchiment. En microscopie à fluorescence, des mesures sont souvent prises pour réduire la quantité de photoblanchiment subie lors de l’expérimentation. Certaines mesures comprennent une réduction de l’intensité de la lumière interagissant avec l’échantillon et l’utilisation de colorants fluorescents spécialisés qui ne restent pas actifs aussi longtemps que les autres colorants.
Microscopie à fluorescence
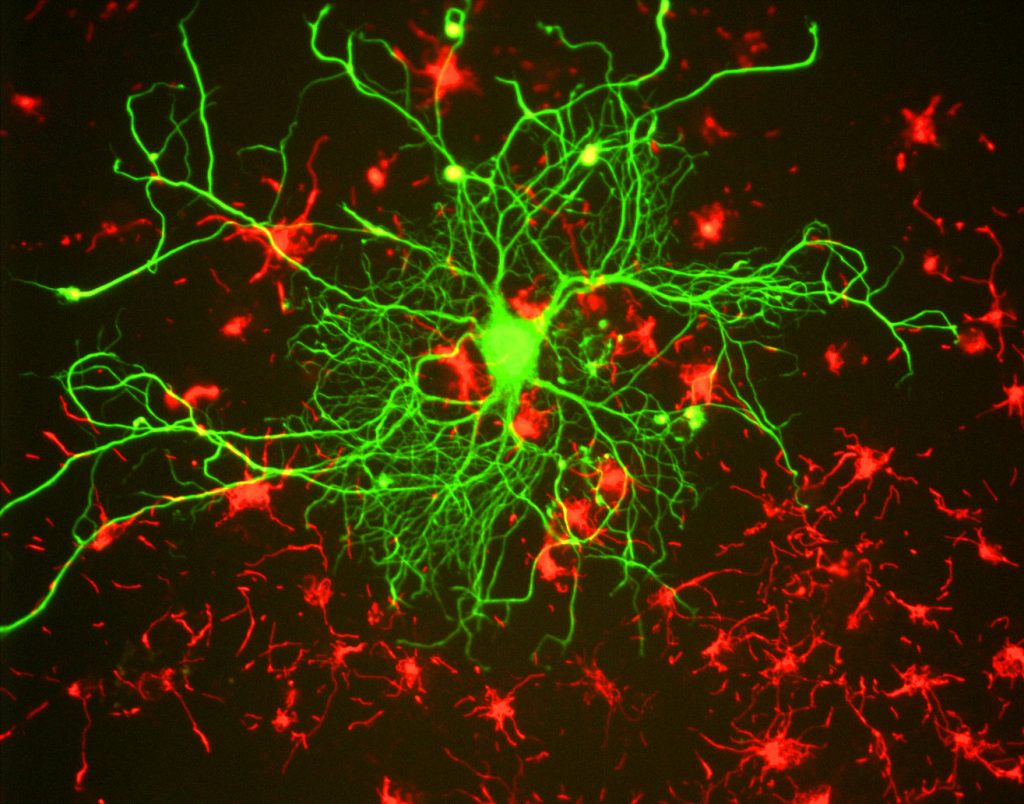
Pour les chercheurs, le principal avantage de la fluorescence est la possibilité d’utiliser la microscopie à fluorescence, où les échantillons sont marqués / colorés avec une substance fluorescente telle qu’un colorant, un anticorps ou une protéine, permettant aux images d’avoir un contraste. En ciblant ces étiquettes fluorescentes, les chercheurs peuvent sélectionner ce qu’ils veulent voir. Ceci est démontré à la Fig.3, où un neurone peut être clairement vu parmi les astrocytes, car ils sont marqués avec différentes couleurs de marqueur fluorescent.
En général, pour la microscopie à fluorescence, un échantillon est marqué avec des marqueurs fluorescents (typiquement spécifiques à certaines parties de l’échantillon). L’échantillon est ensuite éclairé avec la longueur d’onde d’excitation spécifique pour le fluorophore, et la fluorescence d’émission résultante est reçue par le détecteur, généralement une caméra scientifique sensible.
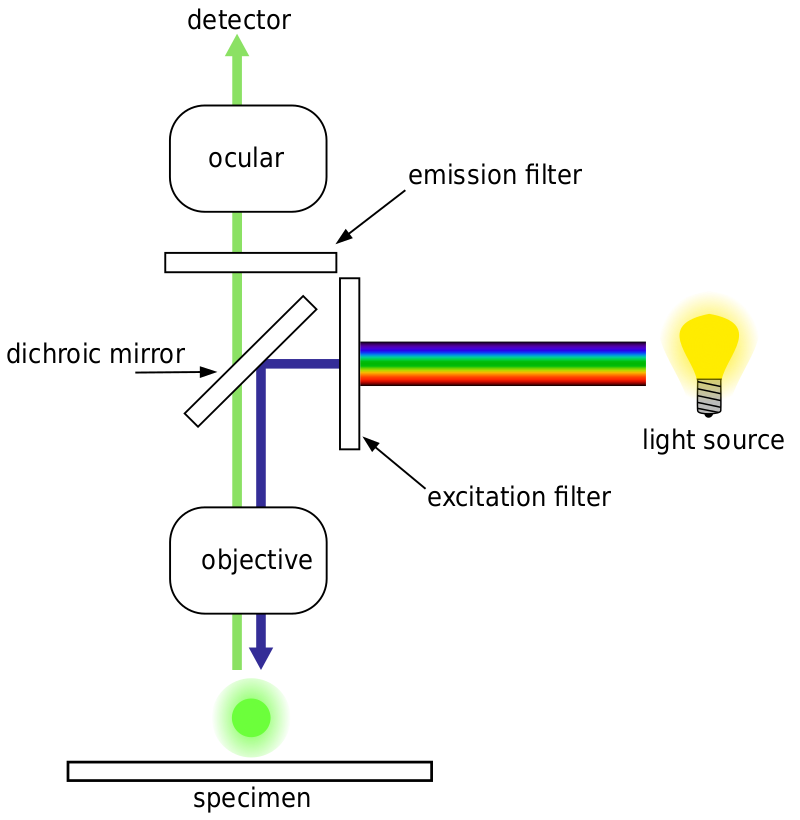
La plupart des microscopes à fluorescence sont des microscopes à épifluorescence, où l’excitation et l’émission se font par le même chemin lumineux. L’illumination d’excitation et la fluorescence émise traversent l’objectif du microscope et sont généralement filtrées afin de détecter simplement la fluorescence. Cette configuration est illustrée à la Fig.4.
Autofluorescence
Certaines structures, organismes biologiques et échantillons de microscopie générale peuvent présenter naturellement une fluorescence, connue sous le nom d’autofluorescence. Ceci est différent de la fluorescence des échantillons marqués, mais il partage souvent des longueurs d’onde similaires, ce qui signifie que les échantillons de microscopie à autofluorescence peuvent masquer la fluorescence artificiellement ajoutée et interférer avec la détection, réduisant le signal. Il est important de savoir si vos échantillons présentent une autofluorescence, car cela affectera toute imagerie de fluorescence effectuée à moins que des longueurs d’onde spécifiques ne soient utilisées pour l’éviter.
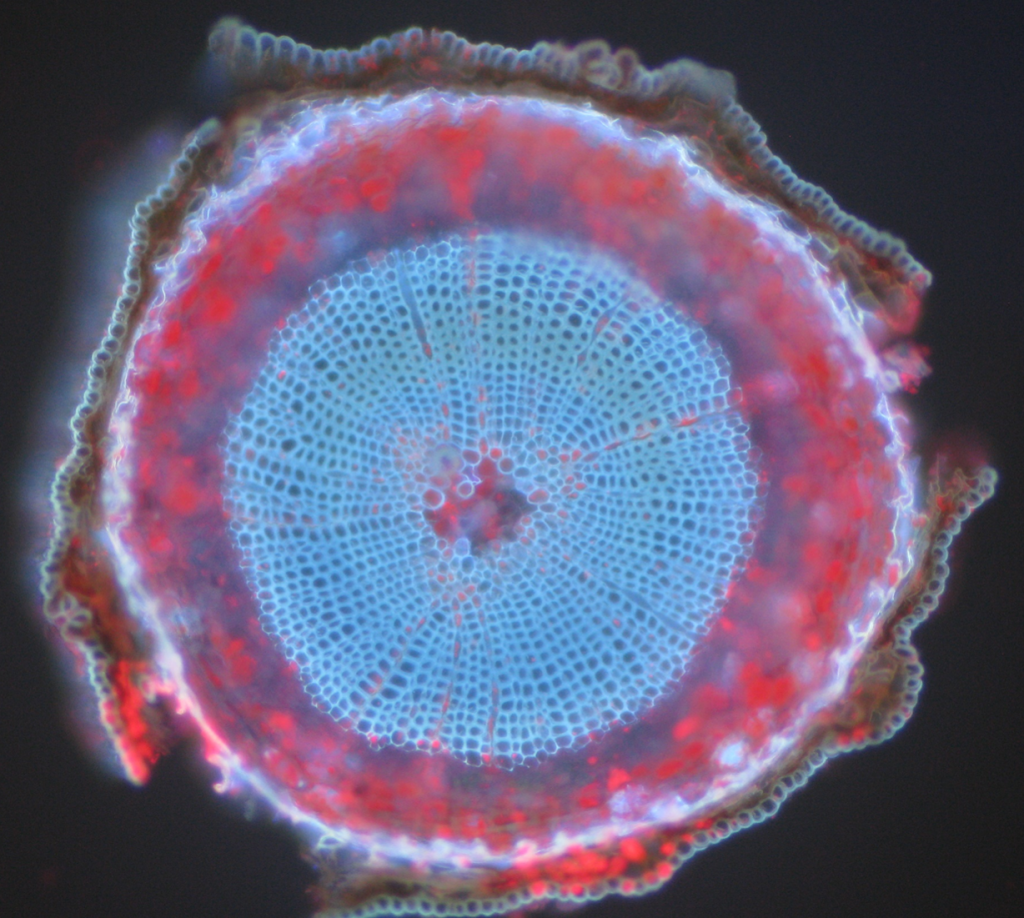
Des exemples courants d’objets autofluorescents sont les mitochondries, les lysosomes, le collagène et certains acides aminés tels que le tryptophane, la tyrosine et la phénylalanine. Plus particulièrement, l’autofluorescence est courante chez les plantes en raison de leur utilisation de chlorophylle et d’autres molécules fluorescentes comme les lignines et les carotènes. Figue.5 montre les différentes couleurs d’autofluorescence d’un échantillon de pin sylvestre non marqué.
Résumé
Depuis l’introduction du premier colorant fluorescent, la microscopie à fluorescence a été un outil très utilisé pour visualiser les cellules et les structures cellulaires avec une plus grande spécificité que les techniques traditionnelles de microscopie à fond clair. Les chercheurs peuventmanipuler la structure, les propriétés optiques et sonder les expériences d’influorescence d’intérêt pour obtenir des données pertinentes. Une telle flexibilité a permis d’inclure la microscopie à fluorescence dans de nombreuses expériences en sciences de la vie.
Selon le type d’échantillon et le fluorophore, une caméra scientifique doit être soigneusement sélectionnée pour obtenir les meilleurs résultats d’imagerie.
- Microscopie optique. (2009) Jalons de la nature. MacMillan Publishers Limited, 6-22.
- Lavis, L. D., & Raines, R.T. (2008). Idées Brillantes pour la Biologie chimique. Biologie chimique ACS, 3 (3): 142-155.
- Liu, Y., Lilly, D. (2017) Structures cristallines si les fluorophores de Cyanine s’empilent à l’extrémité de l’ARN double brin. Biophysical Journal, 113, (11): 2236-2343.
- Berezin, M. Y., Achilefu, S. (2010). Mesures de la Durée de Vie de la Fluorescence et Imagerie Biologique. Revues chimiques, 110 (5): 2641-2684.
- Stockert, J., Blazquez-Castro, A. (2017) Microscopie à fluorescence en sciences de la vie. Sharjah, Émirats arabes Unis. Bentham Science Publishers.
- Berezin, M. Y., & Achilefu, S. (2010). Mesures de la Durée de Vie de la Fluorescence et Imagerie Biologique. Revues chimiques, 110 (5): 2641-2684.
- Denk, W., Strickler, J., Webb, W. (1990). Microscopie à Fluorescence À Balayage Laser À Deux Photons. Sciences. 248, (1951): 73-76.
- Donc, P. (2002). Microscopie optique à fluorescence à Deux photons. Groupe d’édition Macmillan.
- Schermelleh, L., Heinztmann, R. et Leonardt, H. (2010). Un Guide pour la Microscopie à Fluorescence à Super Résolution. Le Journal de biologie cellulaire 190 (2): 165-175.
- Betzig, E., Patterson, G.H., Sougrat, R., Lindwasser, O. W., Olenych, S., Bonifacino, J. S., Davidson, M. W., LippincottSchwartz, J., Hess, H.F. (2006) Imaging intracellular fluorescent proteins at nanometer resolution. Sciences. 313(5793): 1642-5
- Rust, M. J., Bates, M. & Zhuang, X. (2006) Imagerie à limite de sous-diffraction par microscopie de reconstruction optique stochastique (STORM). Méthodes Nat. 2, (10):793-5.
- Rego, E., Shao, L., Macklin, J. Winoto, L., Johansson, G., Kamps-Hughes, N., Davidson, M. et Gustasson, M. (2010) PNAS. 109(3): e135-a143.
- Jungmann, R., Avendaño, M. S., Woehrstein, J. B., Dai, M., Shih, W. M. & Yin, P. (2014) Imagerie cellulaire 3D multiplexée à super-résolution avec PEINTURE à ADN et PEINTURE à échange. Méthodes Nat. 11(3): 313-318
- Jiang, X., Wang, L., Carroll, S., Chen, J., Wang, M. et Wang, J. (2018) Challenges and Opportunities for Small Molecule Fluorescent Probes in Redox Biology Applications. Antioxidants & Redox Signaling. Mary Ann Liebert, Inc.