Differentielle Expressionsanalyse bedeutet, die normalisierten Lesezählungsdaten zu nehmen und statistische Analysen durchzuführen, um quantitative Veränderungen der Expressionsniveaus zwischen experimentellen Gruppen zu entdecken. Zum Beispiel verwenden wir statistische Tests, um zu entscheiden, ob für ein bestimmtes Gen ein beobachteter Unterschied in den Lesewerten signifikant ist, dh ob er größer ist als das, was nur aufgrund natürlicher zufälliger Variation erwartet werden würde.
Methoden zur differentiellen Expressionsanalyse
Es gibt verschiedene Methoden zur differentiellen Expressionsanalyse, wie edgeR und DESeq, die auf negativen Binomialverteilungen (NB) basieren, oder baySeq und EBSeq, Bayes’sche Ansätze, die auf einem negativen Binomialmodell basieren. Es ist wichtig, das experimentelle Design bei der Auswahl einer Analysemethode zu berücksichtigen. Während einige der Tools für Differentialausdrücke nur paarweise Vergleiche durchführen können, können andere wie edgeR, Limma-voom, DESeq und maSigPro mehrere Vergleiche durchführen.
In Abbildung 11 unten skizzieren wir die RNA-seq-Verarbeitungspipeline, die zum Generieren von Daten für Expressionsatlas verwendet wird.
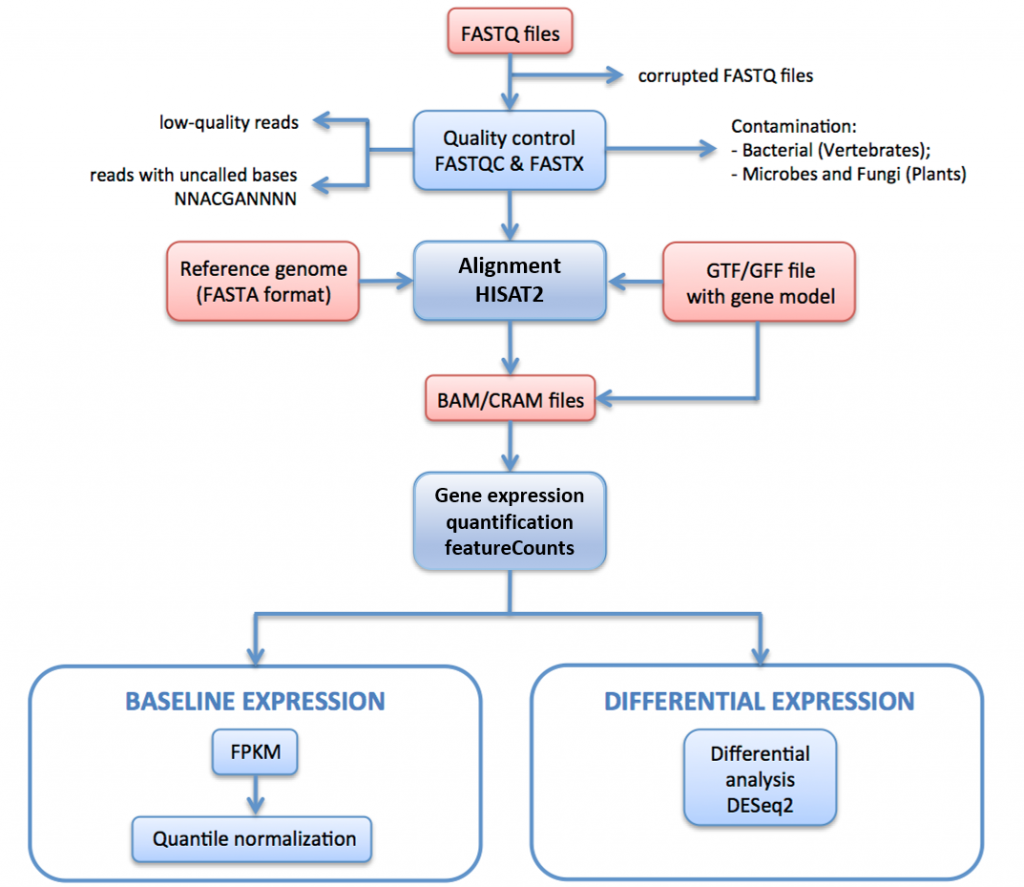
In dieser Pipeline werden rohe Lesevorgänge (FASTQ-Dateien) einer Qualitätsbewertung und Filterung unterzogen. Die qualitätsfilterten Lesevorgänge werden über HISAT2 auf das Referenzgenom ausgerichtet. Die abgebildeten Lesevorgänge werden über HTSeq zusammengefasst und über Gene aggregiert. Für die Baseline-Expression werden die FPKMs von iRAP aus den Rohzählungen berechnet. Diese werden für jeden Satz technischer Replikate gemittelt und dann mit Limma innerhalb jedes Satzes biologischer Replikate quantilnormiert.