Analisi dell’espressione differenziale significa prendere i dati normalizzati del conteggio delle letture ed eseguire analisi statistiche per scoprire cambiamenti quantitativi nei livelli di espressione tra gruppi sperimentali. Ad esempio, usiamo test statistici per decidere se, per un dato gene, una differenza osservata nei conteggi di lettura è significativa, cioè se è maggiore di quanto ci si aspetterebbe solo a causa della variazione casuale naturale.
Metodi per l’analisi dell’espressione differenziale
Esistono diversi metodi per l’analisi dell’espressione differenziale come edgeR e DESeq basati su distribuzioni binomiali negative (NB) o baySeq ed EBSeq che sono approcci bayesiani basati su un modello binomiale negativo. È importante considerare il progetto sperimentale quando si sceglie un metodo di analisi. Mentre alcuni degli strumenti di espressione differenziale possono eseguire solo il confronto a coppie, altri come edgeR, limma-voom, DESeq e maSigPro possono eseguire confronti multipli.
Nella Figura 11, di seguito, descriviamo la pipeline di elaborazione RNA-seq utilizzata per generare dati per Expression Atlas.
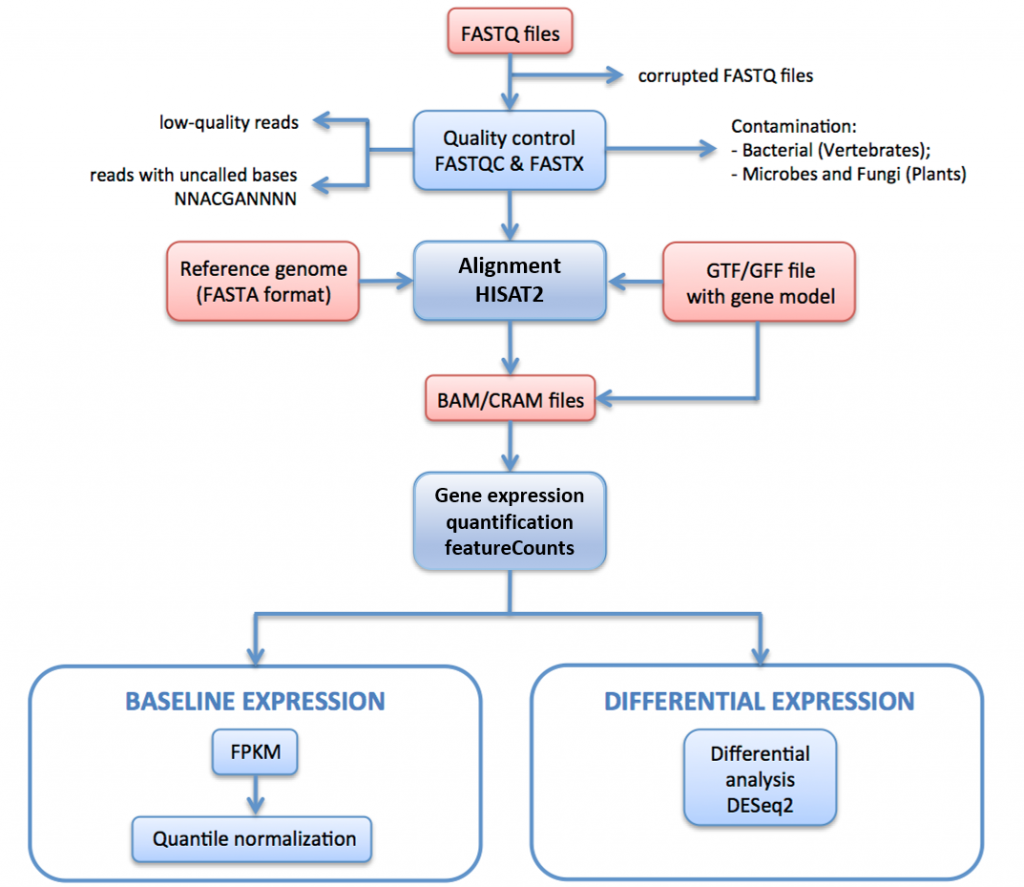
In questa pipeline le letture raw (file FASTQ) sono sottoposte a valutazione e filtraggio della qualità. Le letture filtrate dalla qualità sono allineate al genoma di riferimento tramite HISAT2. Le letture mappate sono riassunte e aggregate su geni tramite HTSeq. Per l’espressione di base, gli FPKM sono calcolati dai conteggi grezzi per iRAP. Questi sono calcolati in media per ogni set di repliche tecniche, e quindi quantili normalizzati all’interno di ogni set di repliche biologiche utilizzando limma.