Fluoreszenzspektroskopie
Die Fluoreszenzspektroskopie wird routinemäßig zur Untersuchung struktureller Veränderungen in konjugierten Systemen, aromatischen Molekülen und starren, planaren Verbindungen aufgrund von Änderungen von Temperatur, pH-Wert, Ionenstärke, Lösungsmittel und Liganden verwendet. Ein einzelner Fluorophor kann Tausende von nachweisbaren Photonen erzeugen, die wiederholt angeregt und nachgewiesen werden können, was die Fluoreszenzspektroskopie zu einer hochempfindlichen Technik macht.
Fluoreszenz ist eine Art Strahlungsemission, die auftritt, wenn ein Molekül Energie bei einer Wellenlänge absorbiert, bei der es ein Übergangsdipolmoment hat. Die Anregungsenergie, die dem Molekül im Grundzustand zur Verfügung gestellt wird, fördert Photonen in einen angeregten Singulettzustand, wo sie dann auf das niedrigste Schwingungsenergieniveau dieses angeregten Singulettzustands zerfallen. Diese Energie entspannt sich weiter zurück in den Grundzustand des Moleküls und emittiert dabei Photonen, wie in Abbildung 1 gezeigt.
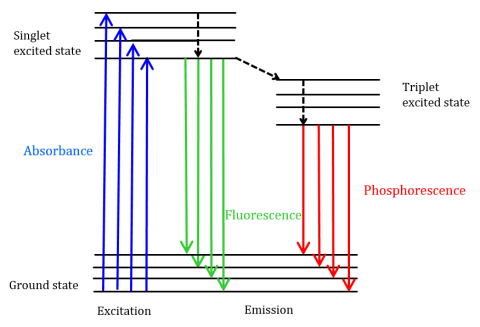
Fluoreszierende Moleküle können sich auch unterziehen Es gibt drei Methoden der nichtstrahlenden Relaxation, bei denen die Anregungsenergie nicht in Photonen umgewandelt wird: (1) interne Umwandlung, (2) externe Umwandlung und (3) Intersystemkreuzung. Eine interne Umwandlung tritt auf, wenn zwischen zwei elektronischen Zuständen eine relativ kleine Energielücke besteht und die Elektronen von einem höheren elektronischen Zustand in einen mit niedrigerer Energie übergehen. Hier wird die Energie auf die Schwingungsmoden des elektronischen Zustands übertragen. Da Schwingungsprozesse thermisch gesteuert werden, führt eine steigende Temperatur zu einer Abnahme der Fluoreszenzintensität. Bei der externen Umwandlung geht Energie durch kollisionales Abschrecken mit gelösten Molekülen in der Umgebung des Fluorophors verloren. Intersystemüberquerung entsteht, wenn sich die Schwingungsniveaus der angeregten Singulett- und Triplettzustände in der Energie überlappen und Elektronen vom niedrigsten angeregten Singulettzustand in den ersten angeregten Triplettzustand übergehen. Die Photonen, die emittiert werden, wenn sie in den Grundzustand zurückkehren, werden als Phosphoreszenz bezeichnet (Abbildung 1). Der Triplett-Zustand hat eine geringere Energie als der Singulett-Zustand, so dass Phosphoreszenz-Peaks bei längeren Wellenlängen als Fluoreszenz gefunden werden. Da diese Übergänge ebenfalls verboten sind, zeigt die Phosphoreszenz eine längere Lebensdauer (~ 10-4 – 102 Sekunden) im Vergleich zur Fluoreszenz (~ 10-9 – 10-6 Sekunden). Die längeren Lebensdauern führen auch zur thermischen Deaktivierung durch Sauerstoffabschreckung, Lösungsmittelbewegung und intermolekulare Kollision, so dass Phosphoreszenz typischerweise nicht bei Raumtemperatur beobachtet werden kann und Proben daher bei Flüssigstickstofftemperatur gekühlt werden müssen.
Biergesetz und Konzentrationseffekte
Während die Absorption auf der Zeitskala von weniger als 10-15 Sekunden erfolgt, ist der Relaxationsprozess vom angeregten in den Grundzustand viel langsamer. Daher kann Fluoreszenz im Gegensatz zur Absorption Informationen über die Wechselwirkungen eines Fluorophors mit umgebenden Molekülen und Lösungsmitteln liefern.
Fluoreszenz intensität ist direkt proportional zu der anregung licht intensität
F = 2,303 * K * I0 * ebc
wo K ist eine konstante basierend auf instrument geometrie, I0 ist die intensität der anregung licht, e ist die fluorophor der molaren absorptionsfähigkeit, b ist die pathlength, und c ist die konzentration. Da die Fluoreszenzintensität nicht wie bei Absorptionsmessungen zur einfallenden Lichtintensität proportional ist, ist die Fluoreszenzempfindlichkeit viel größer, da sie nicht durch die Fähigkeit der Instrumente begrenzt ist, zwischen der einfallenden und der detektierten Intensität zu unterscheiden. Folglich sind kleinere Konzentrationen für Messungen erforderlich.
Die obige Gleichung ist nur linear, wenn die Probenabsorption kleiner als 0,05 AU ist. Wenn eine Probe zu konzentriert ist, kann das Emissionslicht vom Fluorophor reabsorbiert werden, wodurch das Fluoreszenzsignal bei kürzeren Wellenlängen abgeschwächt wird. Anregungslicht kann auch nicht vollständig die gesamte Breite einer hochkonzentrierten Probe durchdringen, was ebenfalls zu verringerten Fluoreszenzintensitäten führt.
Instrumentierung der Fluoreszenzspektroskopie
Eigenschaften eines Fluoreszenzspektrums
Fluorometer bestehen aus einem Anregungs- und Emissionsmonochromator, mit dem Benutzer sowohl Anregungs- als auch Emissionsspektren erhalten können. Eine Messung mit einem Fluorometer ist einzigartig für die Anregungs- und Emissionsmonochromatoren des einzelnen Instruments. Fluoreszenz steht in direktem Zusammenhang mit dem Lichtstrom und der Effizienz der Messung und ist daher vom Gerätedesign und den Komponenten wie Lichtquelle, Monochromatoroptik und Photomultiplier-Röhre abhängig. Jede Lichtquelle hat eine andere spektrale Leistung (sowohl Form als auch Leistung), die über die Lebensdauer der Quelle variiert und abnimmt.
Anregungsspektren zeichnen die Intensität bei einer festen Emissionswellenlänge unter Variation der Anregungswellenlängen auf. Da die meisten Emissionsspektren unabhängig von der Anregungswellenlänge sind, sind die Anregungsspektren häufig Duplikate des Absorptionsspektrums des Fluorophors.
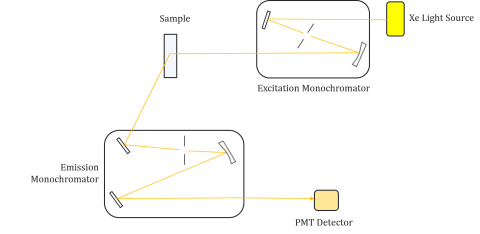
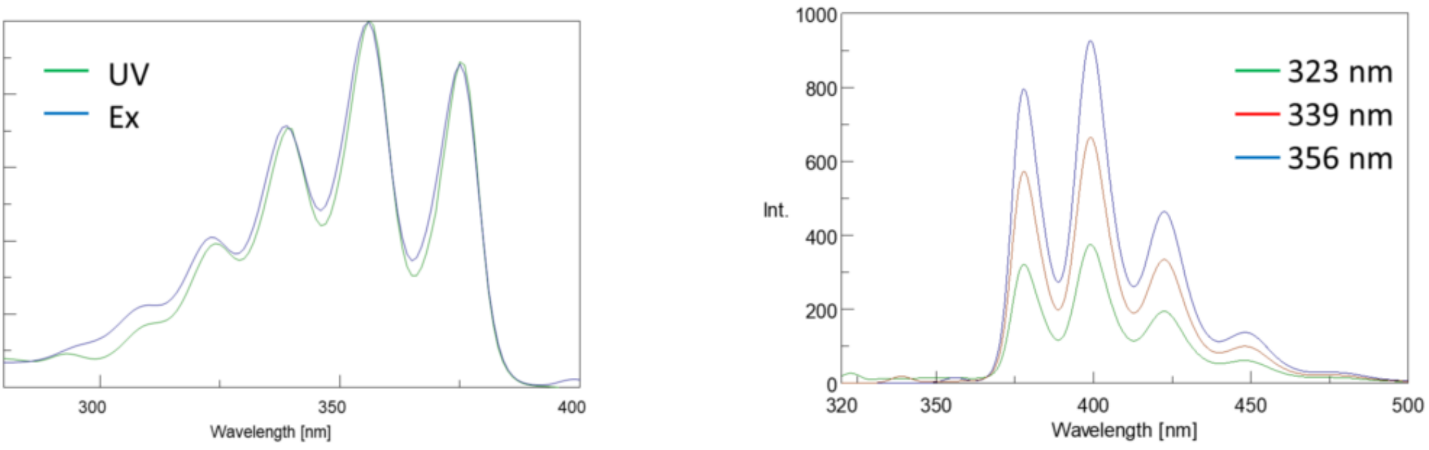
Umgekehrt zeigt ein Emissionsspektrum die Intensität bei einer festen Anregungswellenlänge beim Abtasten durch variierende Emissionswellenlängen. Diese Emissionsscans liefern Informationen über die molekulare Struktur des Fluorophors und die ihn umgebende lokale Umgebung. Da die Fluoreszenzemission immer vom niedrigsten angeregten Zustand bis zum Grundzustand erfolgt, ist die Form des Emissionsspektrums unabhängig von der Anregungswellenlänge. Es wird auch mehr Energie benötigt, um ein Molekül vom Boden in den angeregten Zustand anzuregen, was zu Emissionsspitzen bei längeren Wellenlängen (dh kleineren Energien) als ihren entsprechenden Anregungswellenlängen führt. Dieser Energieunterschied zwischen Anregungs- und Emissionswellenlänge wird als Stokes-Verschiebung bezeichnet.
Darüber hinaus sind Absorptions- und Emissionsspektren aufgrund der gleichen Verteilung zwischen den Schwingungsenergieniveaus des angeregten Zustands und des Grundzustands häufig Spiegelbilder voneinander (Abbildung 3). Das Franck-Condon-Prinzip erklärt, dass, weil die Kerne relativ groß sind und der elektronische Übergang, der an Emission und Absorption beteiligt ist, auf solch schnellen Zeitskalen auftritt, es keine Zeit für Kerne gibt, sich zu bewegen und die Schwingungsenergieniveaus und bleiben daher während des gesamten elektronischen Übergangs ungefähr gleich.
Spektrale Bandbreite
Da die Fluoreszenzintensität proportional zur Eingangslichtintensität ist, beeinflusst die Lichtmenge, die durch den Monochromator geleitet wird, die Intensität stark. Die Summe der Anregungs- und Emissionsbandbreiten sollte etwa der spektralen Bandbreite (SBW) des zu überwachenden Peaks entsprechen, damit alle Peaks gut aufgelöst sind. Solange diese Faustregel befolgt wird, können die Bandbreiten geöffnet werden, um den Lichtdurchsatz für Proben mit geringer Fluoreszenz zu erhöhen. Die SBW kann auch durch die Stokes-Verschiebung des Fluorophors beeinflusst werden. Engere Stokes-Verschiebungen können den Bereich akzeptabler SBWs einschränken, die verwendet werden können.
Fluoreszenzartefakte
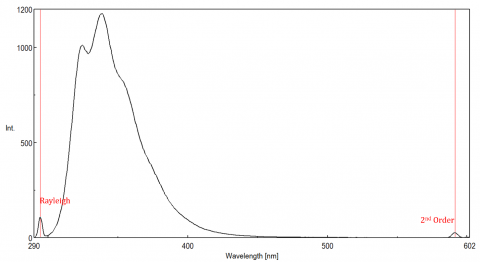
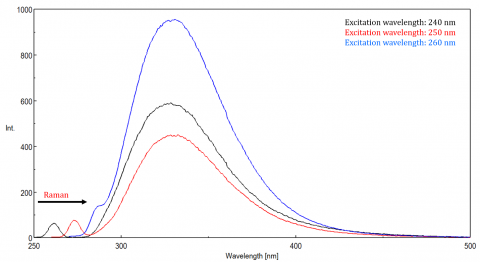
Streulicht kann zu Artefakten führen, die das Fluoreszenzspektrum verzerren. Die drei häufigsten Arten von Streuung in der Fluoreszenz sind Rayleigh, 2. Ordnung und Raman-Streuung (Abbildung 3). Rayleigh-Streuung ist das gestreute Anregungslicht und erreicht daher Spitzenwerte bei der Anregungswellenlänge. Streuung 2. Ordnung ist Streuung höherer Ordnung, die bei der doppelten Anregungswellenlänge beobachtet wird. Raman-Streuung ist eine unelastische Streuung aufgrund von Lösungsmitteln und Peaks bei einer festen Energie aus der Anregungswellenlänge. Um die Raman-Streuung von einem Fluoreszenzpeak zu unterscheiden, kann die Anregungswellenlänge in Schritten von 5 bis 10 nm variiert werden, und wenn sich der betreffende Peak mit der Anregungswellenlänge verschiebt und in der Intensität abnimmt, dann ist dieser Peak auf Raman-Streuung zurückzuführen. Sie können auch überprüfen, ob der Peak im leeren Lösungsmittelspektrum liegt. Wenn ja, besteht die Möglichkeit, dass es sich um einen Raman-Peak handelt. Wenn der Fluoreszenzpeak zu nahe ist oder sich entweder mit der Raman- oder der Rayleigh-Streuung überlappt, können die Bandbreiten und / oder die Anregungswellenlänge angepasst werden, um die Streuung vom Fluoreszenzpeak zu verschieben. Diese Effekte sind für sehr niedrige Fluorophorkonzentrationen und besonders in hohem Grade streuende Lösungen, wie Proteine, Mikrosphäre, nanoparticles sowie Körper am deutlichsten.
Dynamikbereich
Die automatische Verstärkungsregelung passt die Verstärkung eines Signals vom Detektor automatisch an die Fluoreszenzintensität an. Dies optimiert das Signal-Rausch-Verhältnis über den gesamten abgetasteten Bereich für spektrale oder Zeitverlaufsmessungen, so dass Peaks mit unterschiedlichen Intensitäten automatisch angepasst werden, um die S / N zu verbessern und die Ergebnisgenauigkeit zu gewährleisten.
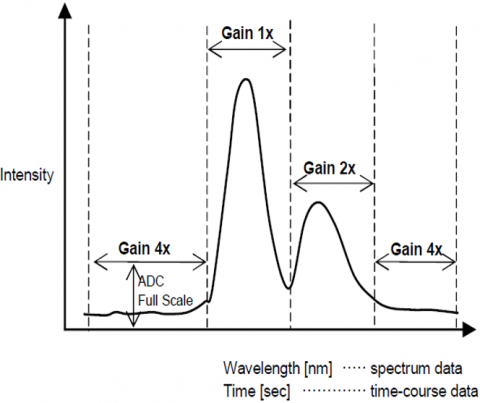
Automatisches Empfindlichkeitsregelungssystem (SCS)
Das automatische Empfindlichkeitsregelungssystem (SCS) erweitert den Dynamikbereich des detektierten Fluoreszenzsignals, indem die Detektorspannung automatisch an die Fluoreszenzintensität angepasst wird. Dies ermöglicht Messungen mit fester Wellenlänge oder quantitativen Analysen von subpikomolaren bis mikromolaren Konzentrationen, ohne das Gerät manuell wechseln zu müssen.

Abbildung 5. Kalibrierungskurve von Fluoresceinlösungen von 5 · 10-13 bis 1,5 · 10-6 M mit der Auto-SCS-Funktion.
Anwendungen der Fluoreszenzspektroskopie
Anisotropie
Fluoreszenz-Anisotropie wird beobachtet, wenn ein Fluorophor Licht unterschiedlicher Intensität in Abhängigkeit von den Polarisationsachsen emittiert und durch die folgende Gleichung beschrieben wird
r=Ivv-GIvh/Ivv+2GIvh
wobei die Emissionsintensität parallel zur Anregungsebene ist und ist die Emissionsintensität senkrecht zur Anregungsebene. G wird als G-Faktor oder Instrumentengitterfaktor bezeichnet und erklärt die Polarisationsabhängigkeit des Emissionsmonochromators.
Alle Fluorophore haben Übergangsmomente, die entlang bestimmter Richtungen entlang der Molekülachse auftreten. Wenn sie polarisiertem Licht ausgesetzt werden, werden die zufällig orientierten Fluorophore, deren Absorptionsübergangsmomente um den Winkel des einfallenden Lichts orientiert sind, angeregt, und diese angeregte Zustandspopulation ist teilweise orientiert. Wenn ein Molekül aus einem angeregten Zustand in seinen Grundzustand zurückkehrt, wird die Elektronenladung umverteilt und die Änderung der Orientierung der Dipolmomente wirkt sich auf die Anregungs- und Emissionspolarisation aus. Wenn beispielsweise Fluoreszenz emittiert wird, bevor sich ein Molekül dreht, wird das Fluoreszenzlicht stark in Richtung der Polarisation des Anregungslichts polarisiert. Wenn das Licht nach der Rotation des Moleküls in eine völlig zufällige Richtung emittiert wird, wird die Fluoreszenz nicht mehr polarisiert.
Bei der Messung der Fluoreszenzanisotropie beeinflussen die folgenden Faktoren die Molekülbewegung: (1) Molekülgröße, (2) Viskosität der Molekülumgebung und (3) Stärke und Freiheitsgrade eines gebundenen Moleküls. Anisotropiemessungen bestimmen die durchschnittliche Winkelverschiebung eines Fluorophors, die zwischen der Absorption und Emission eines Photons auftritt. Die Winkelverschiebung ist abhängig von der Geschwindigkeit und dem Ausmaß der Rotationsdiffusion während der Lebensdauer des angeregten Zustands. Wenn ein Fluorophor uneingeschränkt ist und sich frei drehen kann, bevor ein Photon erneut emittiert wird, ist die Diffusionsrate im Allgemeinen schneller als die Emissionsrate und die Anisotropie ist ungefähr gleich Null. Rotationsdiffusion ändert die Richtung des Übergangsmoments, das die Emission depolarisiert. Je eingeschränkter der Fluorophor ist, desto größer ist der Anisotropiewert, da die Abnahme der Flexibilität die Gesamtrotationsrate verringert.
FRET
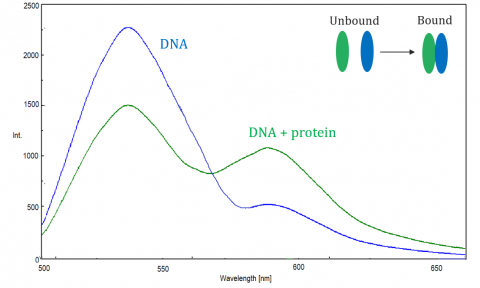
Der Fluoreszenz-Resonanz-Energietransfer (FRET) ist ein Mechanismus, der den Energietransfer zwischen zwei benachbarten Molekülen regelt. Ein Donor, anfänglich in seinem angeregten Zustand, kann Energie durch nichtstrahlende Elektronenresonanz auf ein Akzeptormolekül übertragen.
FRET wird mit dem Spektrofluorometer überwacht, das die Fluoreszenz / Quenchung von Akzeptor oder angeregtem Donor misst. FRET Effizienz hängt von den folgenden Faktoren ab: der Abstand zwischen Donor und Akzeptor, die spektrale Überlappung zwischen Donor und Akzeptor und die Ausrichtung ihrer Dipolmomente. Die Effizienz ist umgekehrt proportional zur sechsten Potenz des Abstands zwischen Donor und Akzeptor, wodurch die Technik extrem empfindlich auf kleine Abstandsänderungen reagiert. Wenn der Überlappungsbereich des Donorfluoreszenzspektrums und des Akzeptorabsorptionsspektrums größer ist, ist die FRET-Effizienz höher. Der FRET-Wirkungsgrad ist auch dann maximal, wenn die beiden Dipolmomente parallel oder antiparallel zueinander sind, und es findet keine Energieübertragung statt, wenn die Dipolmomente senkrecht zueinander stehen. Typischerweise, wenn der Abstand zwischen Donor und Akzeptor zwischen 1 und 10 nm liegt, tritt FRET auf.
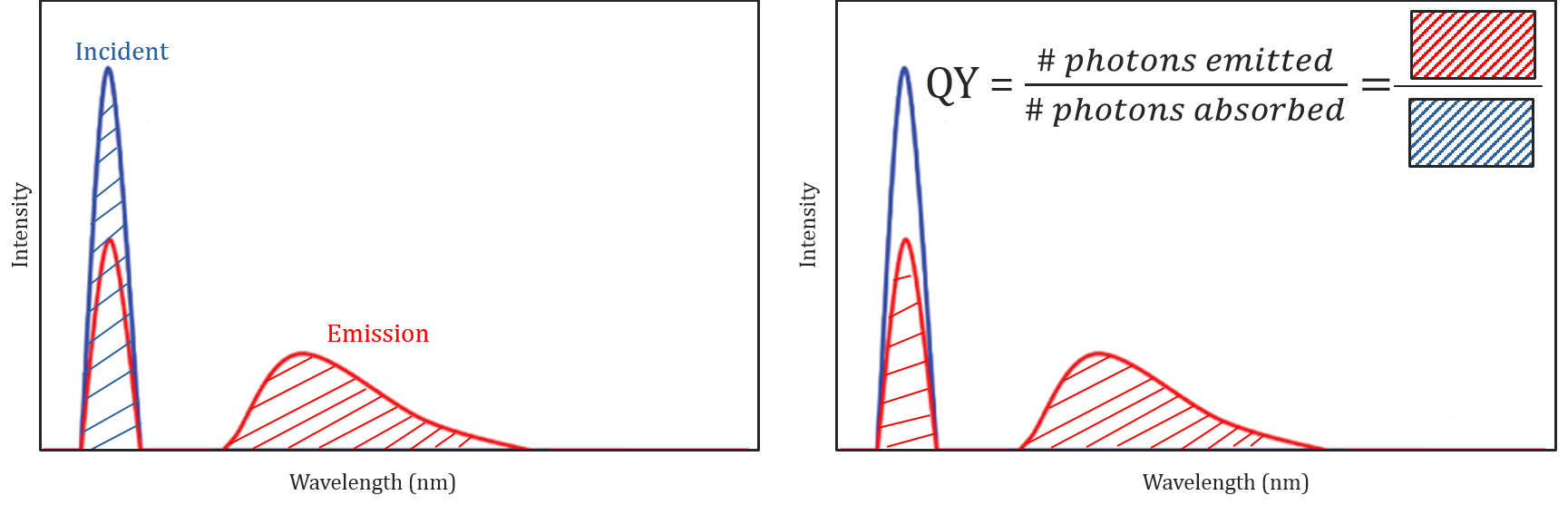
Quantenausbeute und spektrale Korrektur
Unterschiedliche Molekül- und Umgebungsbedingungen beeinflussen nicht nur, ob ein Molekül fluoresziert oder nicht, sondern können auch die Intensität oder Quantenausbeute der emittierten Fluoreszenzstrahlung bestimmen. Die Fluoreszenzeffizienz eines Moleküls wird durch seine Quantenausbeute beschrieben und ist definiert als das Verhältnis der Anzahl der absorbierten Photonen zur Anzahl der von der Probe emittierten Photonen.
In einigen Fällen ist es notwendig, eine genaue spektrale Messung zu bestimmen. Dies erfolgt unter Bezugnahme auf bekannte kalibrierte Materialien. Die verwendeten kalibrierten Quellen werden auf absolute spektrale Leistung an einem bekannten Gerät überprüft und ein Referenzspektrum zur Korrektur des dem Kunden gelieferten Einzelgeräts geliefert. Damit die Spektralkorrektur effektiv funktioniert, muss sie bei jedem Geräteparameter und jeder Bandbreitenkombination durchgeführt werden, sodass die Spektralkorrektur bei einer Spektralbandbreite von 5 nm nicht auf Messungen mit einem 10-nm-SBW angewendet werden kann. Dies gilt für die Position von Polarisatoren, wenn sie verwendet werden, sowie für die Verwendung von Filtern höherer Ordnung. Die spektrale Korrektur muss für jede vom Kunden zu verwendende Kombination von spektralen Bandbreiten, für die Aufnahme oder den Ausschluss der Filterauswahl höherer Ordnung und für die Positionen von Polarisatoren, falls vorhanden, durchgeführt werden. Die Anregungs- und Emissionswellenlängen der Probe bestimmen, welche Lösung / Lichtquelle für die Kalibrierung verwendet wird.
Nahinfrarot
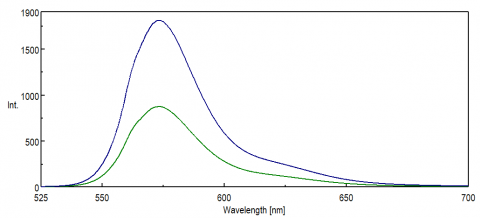
Für Anwendungen, die den NIR-Bereich des Spektrums untersuchen, ist die spektrale Antwort des PMT-Detektors entscheidend für die Datengewinnung. Im roten Ende des sichtbaren Bereichs, im NIR, nimmt die Quanteneffizienz des PMT signifikant ab, was zu einer geringen bis gar keiner Signalintensität bei Probenmessungen führt. FRET-Experimente und NIR-Farbstoffe und -Sonden werden häufig bei Wellenlängen über 500 nm überwacht und haben in vielen Fällen kleine Signale, selbst für eine so empfindliche Technik wie Fluoreszenz. Abbildung 8 veranschaulicht den Unterschied in der Fluoreszenzintensität von Rhodamin B unter Verwendung einer Standard-PMT im Vergleich zu einer PMT, die empfindlicher für Wellenlängen am roten Ende des Spektrums ist.
Komplementäre Technik:
Zirkulardichroismusspektroskopie
Die Zirkulardichroismusspektroskopie (CD) ist eine wesentliche Analysetechnik zur Analyse der Chiralität in Molekülen durch ihre optische Aktivität. CD kann auf eine Vielzahl von molekularen Strukturen angewendet werden, hat aber in der wissenschaftlichen Gemeinschaft für die Aufklärung der makromolekularen Struktur, insbesondere Proteine und Nukleinsäuren, Gefallen gefunden.