introduktion
væv, celler og de mindre strukturer inde i celler (organeller) er for det meste vand og er derfor gennemsigtige. Billeddannelse af små gennemsigtige poser med vand resulterer i billeder, der ikke indeholder meget information, og i mikroskopi er det vigtigt at have en slags kontrast eller plet, der giver områder af prøvefarven og gør dem langt lettere at se. Derudover, hvad hvis du kun vil afbilde nogle af de mindre strukturer inde i en celle, som en kerne eller en cellemembran? Farvning af hele cellen ville gøre det umuligt at lokalisere de områder, du er interesseret i.
fluorescens løser begge disse problemer med kontrast og lokalisering. Fluorescens er, hvor et objekt udsender lys efter at have absorberet lys. Mange forskellige genstande udviser fluorescens, såsom mineraler (ordet fluorescens kommer fra mineralet fluorit), dybhavsfisk (mest berømt vandmænd Aekvorea victoria, hvorfra grønt fluorescerende protein (GFP) blev opdaget), planter, kemikalier og mange flere.
fluorescerende molekyler (kendt som fluoroforer) bruges til at mærke prøver, og fluoroforer er tilgængelige, der udsender lys i stort set enhver farve. I et fluorescerende mikroskop mærkes en prøve med en fluorofor, og derefter bruges et stærkt lys (eksitationslys) til at belyse prøven, som afgiver fluorescens (emissionslys). På denne måde er prøver stærkt kontrast til den sorte baggrund, da fluoroforen udsender et lyst farvet lys. Ved at lokalisere disse fluoroforer til interesseområdet kan der tages et klart billede af enhver del af en celle, hvilket gør fluorescensmikroskopi til et kraftfuldt værktøj til biovidenskab.
Brightfield vs Fluorescensafbildning
i brightfield-mikroskopi lyser prøven med transmitteret hvidt lys. Dette skaber en jævn belysning af prøven under mikroskopet for at observere stærkt kontrasterede, farvede eller naturligt pigmenterede prøver. Brightfield er imidlertid ikke tilstrækkelig til at skelne mellem gennemsigtige/gennemskinnelige, ufarvede celler eller cellulære strukturer til at studere processer af interesse.
fluorescensmikroskopi er afhængig af brugen af fluoroforer, molekyler, der udsender lys med en specifik synlig bølgelængde, når de udsættes for lys med en anden bølgelængde. Når disse fluoroforer er bundet til en målrettet struktur af interesse, kan fotoner udsendt fra fluoroforen bruges til at visualisere denne struktur af interesse. Fordelen ved fluorescensmikroskopi er, at de målrettede strukturer er belyst, mens de uønskede områder af prøven har ringe eller ingen fluorescens, hvilket giver mulighed for let målretning og billeddannelse.
hvorfor molekyler fluorescerer
oprindelsen af fluorescens er elektroner, der bevæger sig frit omkring den aktive fluorofor og frigiver absorberet energi, som det ses i Fig.2.
før spænding er elektroner i den laveste energitilstand, der er tilgængelig for dem – jordtilstanden (S0). Når en elektron rammes med en foton i et bestemt energiområde, absorberer elektronen fotonens energi og hopper op til en højere energitilstand (S1, S2 eller S3). For at vende tilbage til jordtilstanden (S0) frigiver elektronen den ekstra energi som emission af en foton. Energien i denne foton er mindre end eksitationsenergien, så den har en længere bølgelængde. Dette er grunden til, at emissionslys har en længere bølgelængde end eksitationslyset og kan fremstå som en anden farve.
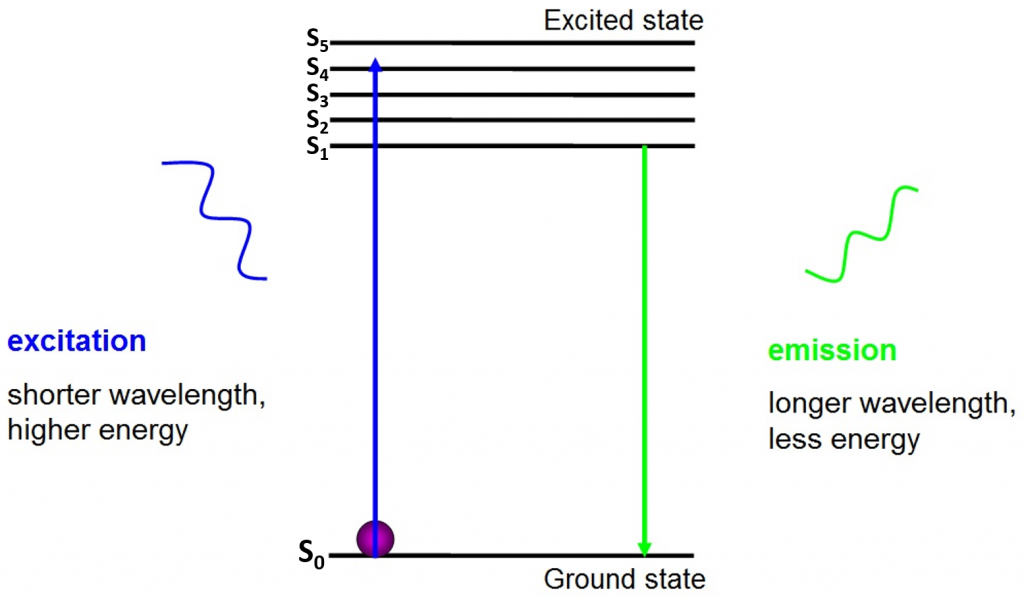
den udsendte foton er normalt i det synlige spektrum og kan ses under et mikroskop, hvis der er nok ophidsede fluoroforer. Bølgelængden af den frigivne foton er specifik for hver fluorofor, og denne forudsigelighed muliggør let fluorescensafbildning.
Fluorescensintensitetsfaktorer
mens fluoroforer kan udsende fluorescens med en forudsigelig bølgelængde, er det også vigtigt at vide, hvilke faktorer der styrer fluorescensintensitet. Uden en intens nok emission af lys kan fluorescensen ikke påvises med et mikroskop.
Kvanteudbytte
kvanteudbyttet (liter) af en fluorofor er forholdet mellem antallet af frigivne fotoner og antallet af absorberede fotoner. Kvanteudbyttet udtrykkes ofte som en værdi fra 0-1, hvoraf 1 er 100% effektivitet af fotonkonvertering. Det er også vigtigt at bemærke, at hver fluorofor har en unik pH, ionstyrke og temperatur for optimal fluorescenseffektivitet.
Udryddelseskoefficient
hver fluorofor har en anden kapacitet til at absorbere fotoner, selvom de er inden for et passende bølgelængdeområde for at begejstre det. Hvis en fluorofor udsættes for en foton, der passer passende til dens eksitationsbølgelængde, er sandsynligheden for, at en foton absorberes, en målbar egenskab og kendt som udryddelseskoefficient (liter).
en fluorophores kvanteudbytte og udryddelseskoefficient vises ofte sammen for at beskrive, hvor lys fluorophoren viser sig at være i eksperimentelle indstillinger.
fluorescens levetid
når en fluoroforelektron absorberer en foton, frigiver den ikke straks en længere bølgelængdefoton. Frigivelsen af noget energi mellem ophidsede energitilstande vides at tage forskellige længder af tiden. Mængden af tid, som en elektron bruger i ophidset tilstand, før han frigiver en foton og vender tilbage til jordtilstanden, er en måling af dens fluorescenslevetid. Levetiden for hver fluorofor er unik og kan måles eksperimentelt. Når du bruger fluorescerende farvestoffer eksperimentelt, er det nyttigt for deres levetid, især til applikationer, der kræver høj hastighed, såsom calciumafbildningsneuroner.
Spændingsbølgelængdeintensitet
de fleste fluorescerende mikroskopiopsætninger inkluderer en lyskilde, der kan indstilles til at udsende det ønskede bølgelængdeområde. Mange fluorescerende lyskilder kan også justeres for eksitationsintensitet for at øge antallet af fotoner, der bevæger sig gennem lysstien. I en fluorescerende mærket prøve, der udsættes for dens eksitationsbølgelængde, vil hver fluorofor ikke blive aktiveret på samme tid. Ved at øge eksitationsintensiteten og øge antallet af fotoner, der når prøven, er der en større sandsynlighed for, at flere fluoroforer vil blive ophidset.
fotostabilitet
fotostabilitet er et molekyles eller organismes evne til at modstå skader. I fluorescensmikroskopi vil fluoroforer til sidst stoppe med at absorbere modkørende fotoner og gå i en permanent mørk tilstand. Når en organisme samler flere fluoroforer i mørk tilstand, reduceres udseendet af det mærkede mål, og prøven siges at være fotoblegning. I fluorescensmikroskopi tages der ofte skridt til at reducere mængden af fotoblegning, der opleves under eksperimentering. Nogle foranstaltninger inkluderer en reduktion i intensiteten af lys, der interagerer med prøven, og brugen af specialiserede fluorescerende farvestoffer, der ikke forbliver aktive så længe som andre farvestoffer.
fluorescensmikroskopi
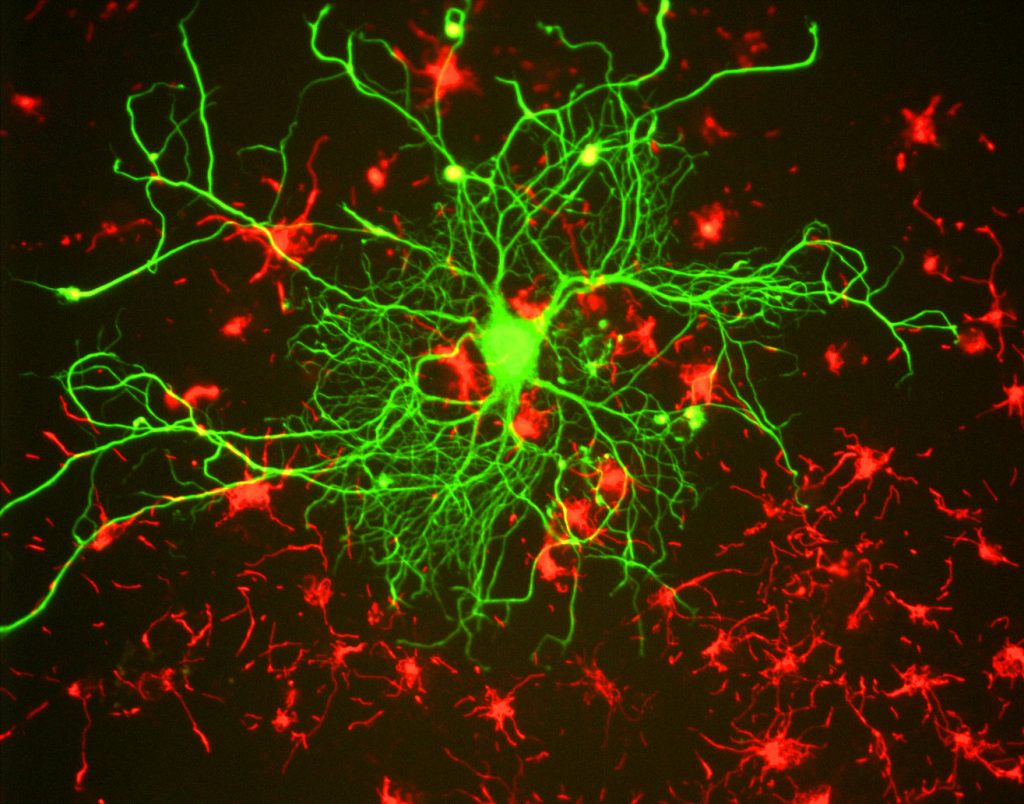
for forskere er den største fordel ved fluorescens evnen til at anvende fluorescensmikroskopi, hvor prøver mærkes/farves med et fluorescerende stof, såsom et farvestof, antistof eller protein, så billeder kan have kontrast. Ved at målrette disse fluorescerende etiketter kan forskere vælge, hvad de vil se. Dette er vist i Fig.3, hvor en neuron tydeligt kan ses blandt astrocytter, da de er mærket med forskellige farver af fluorescerende markør.
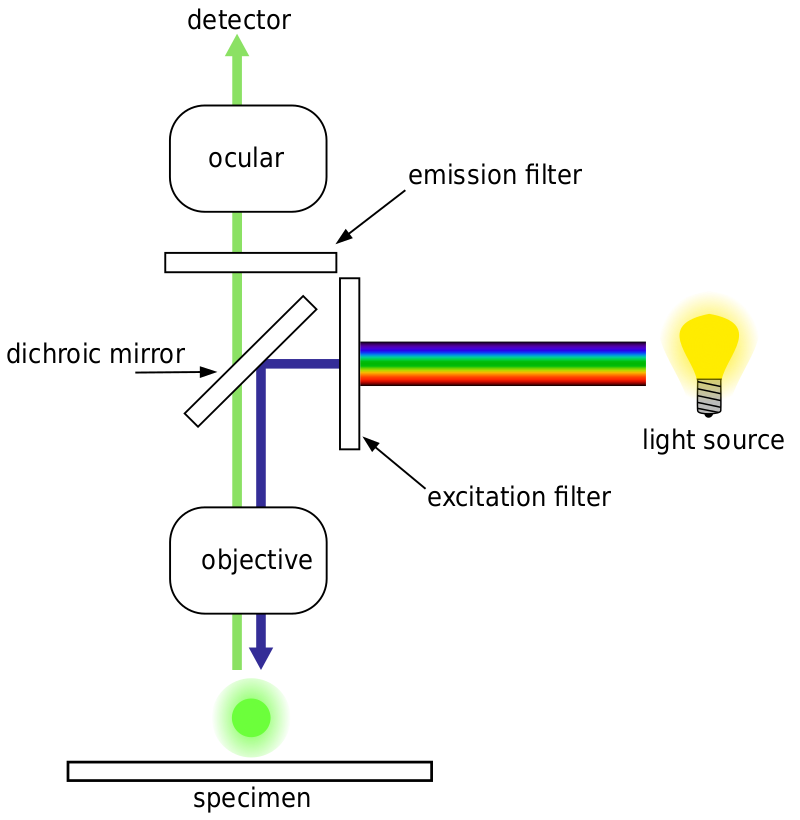
generelt for fluorescensmikroskopi er en prøve mærket med fluorescerende markører (typisk specifik for visse dele af prøven). Prøven belyses derefter med den specifikke eksitationsbølgelængde for fluoroforen, og den resulterende emissionsfluorescens modtages af detektoren, normalt et følsomt videnskabeligt kamera.
de fleste fluorescensmikroskoper er epifluorescensmikroskoper, hvor ophidselse og emission udføres gennem den samme lyssti. Både eksitationsbelysningen og den udsendte fluorescens passerer gennem mikroskopmålet og filtreres normalt for blot at detektere fluorescensen. Denne opsætning er vist i Fig.4.
Autofluorescens
nogle strukturer, biologiske organismer og generelle mikroskopiprøver kan naturligt udvise fluorescens, kendt som autofluorescens. Dette adskiller sig fra fluorescens fra mærkede prøver, men det deler ofte lignende bølgelængder, hvilket betyder, at prøver af autofluorescensmikroskopi kan skjule kunstigt tilsat fluorescens og interfererer med detektion, reducerende signal. Det er vigtigt at vide, om dine prøver udviser autofluorescens, da dette vil påvirke enhver fluorescensafbildning udført, medmindre specifikke bølgelængder bruges til at undgå det.
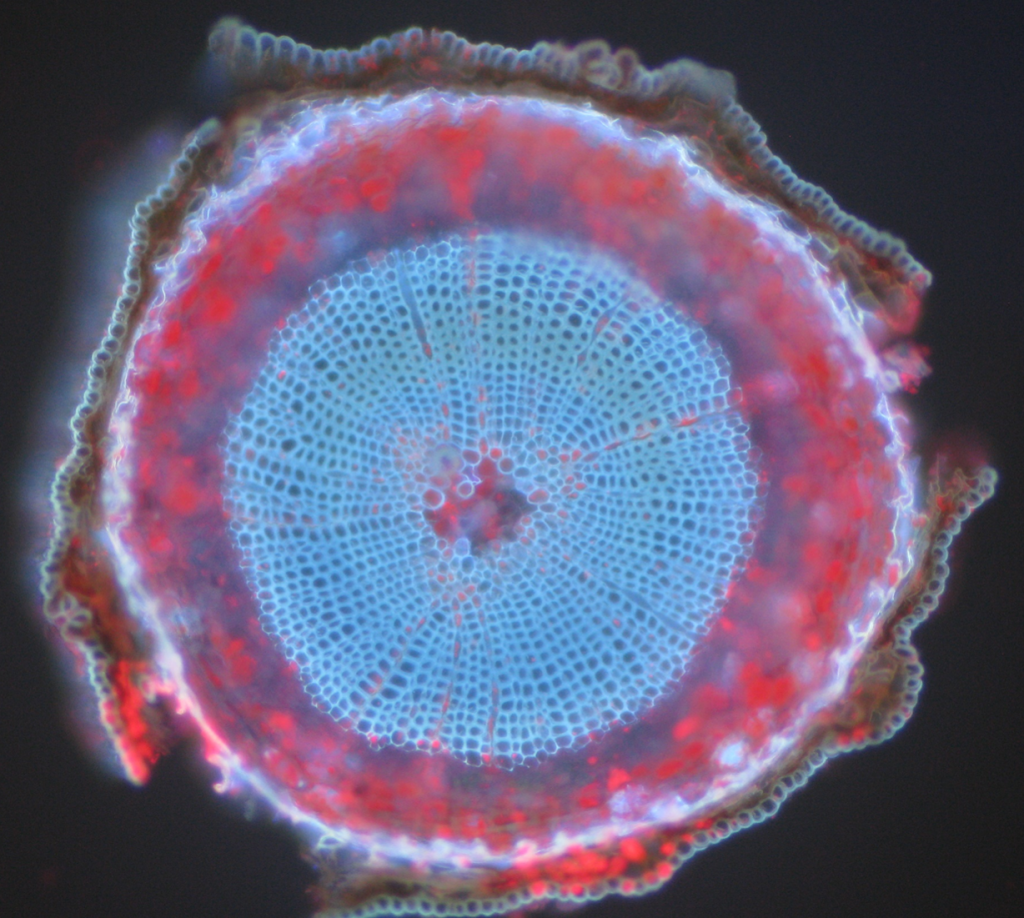
almindelige eksempler på autofluorescerende objekter er mitokondrier, lysosomer, kollagen og nogle aminosyrer såsom tryptophan, tyrosin og phenylalanin. Mest bemærkelsesværdigt er autofluorescens almindelig i planter på grund af deres anvendelse af klorofyl og andre fluorescerende molekyler som ligniner og carotener. Fig.5 viser de forskellige farver af autofluorescens fra en umærket skotsk fyrprøve.
Resume
siden indførelsen af det første fluorescerende farvestof er fluorescensmikroskopi blevet et stærkt anvendt værktøj til at visualisere celler og cellulære strukturer med højere specificitet end traditionelle Brightfield mikroskopiteknikker. Forskere kanmanipulere strukturen, optiske egenskaber og sonde af interesse influorescens eksperimenter for at opnå relevante data. En sådan fleksibilitet har muliggjort, at fluorescensmikroskopi kan indgå i mange life science Eksperimenter.
afhængigt aftype prøve og fluorofor, et videnskabeligt kamera skal omhyggeligt vælges for at opnå de bedste billeddannelsesresultater.
- Lysmikroskopi. (2009) Natur Milepæle. MacMillan Publishers Limited, 6-22.
- Lavis, L. D., & Raines, R. T. (2008). Lyse ideer til kemisk biologi. ACS kemisk biologi, 3 (3): 142-155.
- Liu, Y., Lilly, D. (2017) krystalstrukturer, hvis Cyaninfluorophorer stabler på enden af dobbeltstrenget RNA. Biofysisk Tidsskrift, 113, (11): 2236-2343.
- Beresin, M. Y., Achilefu, S. (2010). Fluorescens Livstidsmålinger og biologisk billeddannelse. Kemiske Anmeldelser, 110 (5): 2641-2684.
- Stockert, J., Castro, A. (2017) fluorescensmikroskopi i biovidenskab. Sharjah, UAE. Bentham Science Publishers.
- Beresin, M. Y., & Achilefu, S. (2010). Fluorescens Livstidsmålinger og biologisk billeddannelse. Kemiske Anmeldelser, 110 (5): 2641-2684.
- Denk, V., Strickler, J., V., V. (1990). To-Foton Laser Scanning Fluorescens Mikroskopi. Videnskab. 248, (1951): 73-76.
- Så, P. (2002). To-foton fluorescens lysmikroskopi. Macmillan Publishing Group.
- Schermelleh, L., Heintsmann, R. og Leonardt, H. (2010). En Guide til fluorescensmikroskopi med Superopløsning. Tidsskriftet for cellebiologi 190 (2): 165-175.
- Betsig, E., Patterson, G. H., Sougrat, R., Lindvasser, O. V., Olenych, S., Bonifacino, J. S., Davidson, M. V., Lippincottschvarts, J., Hess, H. F. (2006) billeddannelse af intracellulære fluorescerende proteiner ved nanometeropløsning. Videnskab. 313(5793): 1642-5
- Rust, M. J., Bates, M. & Chuang, S. (2006) subdiffraktionsgrænsebilleddannelse ved stokastisk optisk rekonstruktionsmikroskopi (STORM). Nat Metoder. 2, (10):793-5.
- Rego, E., Shao, L., Macklin, J. Vinoto, L., Johansson, G., Kamps-Hughes, N., Davidson, M. Og Gustasson, M. (2010) PNAS. 109 (3): e135-a143.
- Jungmann, R., Avenda Larso, M. S., J. B., Dai, M., Shih, M. M. & Yin, P. (2014) multiplekset 3D cellulær superopløsningsbilleddannelse med DNA-maling og Udvekslingsmaling. Nat Metoder. 11(3): 313-318
- J. J., J. J., J. (2018) Challenges and Opportunities for Small Molecule Fluorescent Probes in Redox Biology Applications. Antioxidants & Redox Signaling. Mary Ann Liebert, Inc.