Spectroscopie de fluorescence
La spectroscopie de fluorescence est couramment utilisée pour étudier les changements structurels des systèmes conjugués, des molécules aromatiques et des composés plans rigides dus à des altérations de la température, du pH, de la force ionique, du solvant et des ligands. Un seul fluorophore peut générer des milliers de photons détectables qui peuvent être excités et détectés à plusieurs reprises, ce qui rend la spectroscopie de fluorescence très sensible.
La fluorescence est un type d’émission radiative qui se produit lorsqu’une molécule absorbe de l’énergie à une longueur d’onde où elle a un moment dipolaire de transition. L’énergie d’excitation fournie à la molécule à l’état fondamental favorise les photons à un état singulet excité, où ils se désintègrent ensuite au niveau d’énergie vibratoire le plus bas de cet état singulet excité. Cette énergie se détend encore à l’état fondamental de la molécule, émettant des photons dans le processus, comme le montre la figure 1.
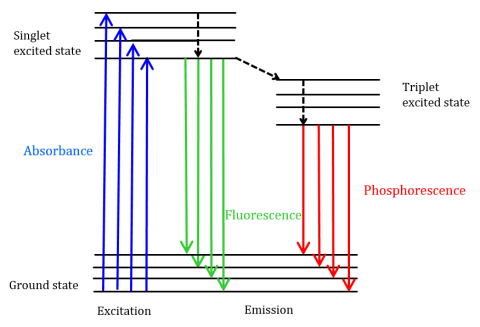
Les molécules fluorescentes peuvent également subir il existe trois méthodes de relaxation non radiative où l’énergie d’excitation n’est pas convertie en photons: (1) conversion interne, (2) conversion externe et (3) croisement entre les systèmes. La conversion interne se produit lorsqu’il existe un écart d’énergie relativement faible entre deux états électroniques et que les électrons passent d’un état électronique supérieur à un état d’énergie inférieure. Ici, l’énergie est transférée aux modes vibratoires de l’état électronique. Étant donné que les processus vibratoires sont entraînés thermiquement, l’augmentation de la température entraîne une diminution de l’intensité de la fluorescence. Dans la conversion externe, l’énergie est perdue par trempe collisionnelle avec des molécules de soluté dans l’environnement du fluorophore. Le croisement d’intersystèmes se produit lorsque les niveaux vibrationnels des états excités singulet et triplet se chevauchent dans la transition d’énergie et d’électrons de l’état excité singulet le plus bas au premier état excité triplet. Les photons émis lorsqu’ils reviennent à l’état fondamental sont connus sous le nom de phosphorescence (figure 1). L’état triplet est plus faible en énergie que l’état singulet, de sorte que les pics de phosphorescence se trouvent à des longueurs d’onde plus longues que la fluorescence. Comme ces transitions sont également interdites, la phosphorescence présente une durée de vie plus longue (~ 10-4 – 102 secondes) par rapport à la fluorescence (~ 10-9 – 10-6 secondes). Les durées de vie plus longues entraînent également une désactivation thermique par trempe à l’oxygène, mouvement du solvant et collision intermoléculaire, de sorte que la phosphorescence ne peut généralement pas être observée à température ambiante et que les échantillons doivent donc être refroidis à la température de l’azote liquide.
Loi de la bière et Effets de concentration
Alors que l’absorption se produit sur une échelle de temps inférieure à 10-15 secondes, le processus de relaxation de l’état excité à l’état fondamental est beaucoup plus lent. Par conséquent, la fluorescence peut fournir des informations sur les interactions des fluorophores avec les molécules et les solvants environnants, contrairement à l’absorption.
L’intensité de fluorescence est directement proportionnelle à l’intensité lumineuse d’excitation
F = 2,303 * K * I0 * ebc
où K est une constante basée sur la géométrie de l’instrument, I0 est l’intensité de la lumière d’excitation, e est l’absorptivité molaire du fluorophore, b est la longueur du trajet et c est la concentration. Comme l’intensité de fluorescence n’est pas rapportée à l’intensité lumineuse incidente comme pour les mesures d’absorption, la sensibilité de fluorescence est beaucoup plus grande car elle n’est pas limitée par la capacité des instruments à différencier les intensités incidentes et détectées. Par conséquent, des concentrations plus faibles sont nécessaires pour les mesures.
L’équation ci-dessus n’est linéaire que lorsque l’absorbance de l’échantillon est inférieure à 0,05 UA. Si un échantillon est trop concentré, la lumière d’émission peut être réabsorbée par le fluorophore, atténuant le signal de fluorescence à des longueurs d’onde plus courtes. La lumière d’excitation peut également ne pas pénétrer complètement sur toute la largeur d’un échantillon hautement concentré, ce qui entraînera également une diminution des intensités de fluorescence.
Instrumentation de la spectroscopie de fluorescence
Caractéristiques d’un spectre de fluorescence
Les fluoromètres sont composés d’un monochromateur d’excitation et d’émission, permettant aux utilisateurs d’obtenir à la fois des spectres d’excitation et d’émission. Une mesure effectuée par un fluoromètre est unique aux monochromateurs d’excitation et d’émission de chaque instrument. La fluorescence est directement liée au flux lumineux et à l’efficacité de la mesure et dépend donc de la conception de l’instrument et de ses composants tels que la source de lumière, l’optique monochromatrice et le tube photomultiplicateur. Chaque source de lumière aura une sortie spectrale différente (à la fois de forme et de puissance) qui variera et diminuera au cours de la durée de vie de la source.Les spectres d’excitation
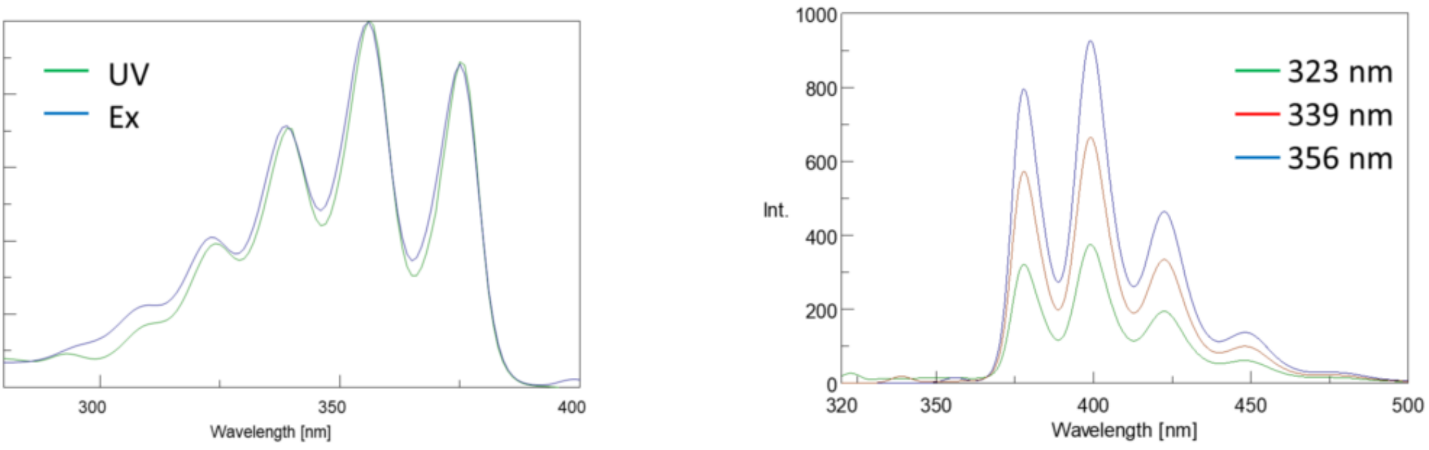
tracent l’intensité à une longueur d’onde d’émission fixe tout en faisant varier les longueurs d’onde d’excitation. Étant donné que la plupart des spectres d’émission sont indépendants de la longueur d’onde d’excitation, les spectres d’excitation sont souvent des doublons du spectre d’absorption du fluorophore.
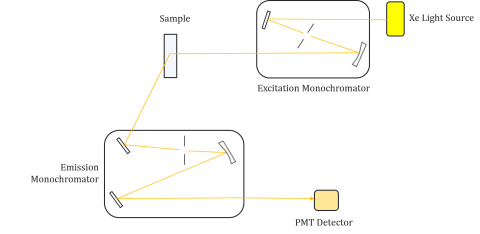
Inversement, un spectre d’émission trace l’intensité à une longueur d’onde d’excitation fixe tout en balayant à travers des longueurs d’onde d’émission variables. Ces analyses d’émission fournissent des informations sur la structure moléculaire du fluorophore et l’environnement local qui l’entoure. Comme l’émission de fluorescence se produit toujours de l’état excité le plus bas à l’état fondamental, la forme du spectre d’émission est indépendante de la longueur d’onde d’excitation. Plus d’énergie est également nécessaire pour exciter une molécule du sol à l’état excité, ce qui entraîne des pics d’émission à des longueurs d’onde plus longues (c’est-à-dire des énergies plus petites) que leurs longueurs d’onde d’excitation correspondantes. Cette différence d’énergie entre les longueurs d’onde d’excitation et d’émission est connue sous le nom de décalage de Stokes.
De plus, les spectres d’absorption et d’émission sont souvent des images miroir l’un de l’autre en raison de la distribution égale entre les niveaux d’énergie vibratoire des états excités et des états fondamentaux (Figure 3). Le principe de Franck-Condon explique que parce que les noyaux sont relativement grands et que la transition électronique impliquée dans l’émission et l’absorption se produit sur des échelles de temps aussi rapides, il n’y a pas de temps pour que les noyaux se déplacent et que les niveaux d’énergie vibratoire restent donc à peu près les mêmes tout au long de la transition électronique.
Bande passante spectrale
Étant donné que l’intensité de fluorescence est proportionnelle à l’intensité lumineuse d’entrée, la quantité de lumière passée à travers le monochromateur affectera considérablement l’intensité. La somme des largeurs de bande d’excitation et d’émission doit correspondre à la largeur de bande spectrale (SBW) du pic surveillé afin que tous les pics soient bien résolus. Tant que cette règle empirique est respectée, les largeurs de bande peuvent être ouvertes pour augmenter le débit de lumière pour les échantillons à faible fluorescence. Le SBW peut également être affecté par le déplacement de Stokes du fluorophore. Des décalages de Stokes plus étroits peuvent limiter la plage de SBW acceptables pouvant être utilisés.
Artefacts de fluorescence
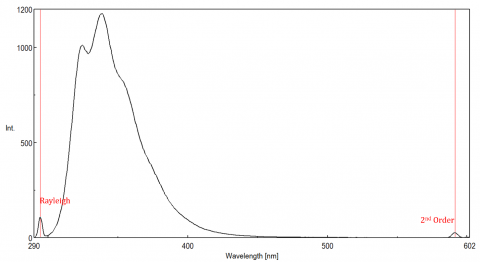
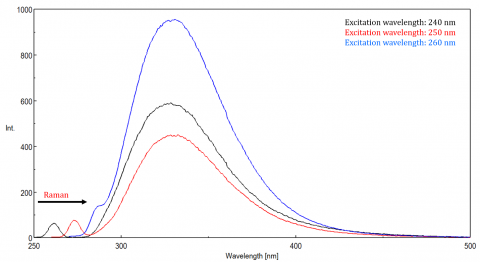
La lumière diffusée peut donner lieu à des artefacts, déformant le spectre de fluorescence. Les trois types de diffusion les plus courants observés en fluorescence sont la diffusion de Rayleigh, du 2e ordre et la diffusion de Raman (Figure 3). La diffusion de Rayleigh est la lumière d’excitation diffusée et donc des pics à la longueur d’onde d’excitation. la diffusion du 2ème ordre est une diffusion d’ordre supérieur observée à deux fois la longueur d’onde d’excitation. La diffusion Raman est une diffusion inélastique due aux solvants et aux pics à une énergie fixe de la longueur d’onde d’excitation. Pour différencier la diffusion Raman d’un pic de fluorescence, la longueur d’onde d’excitation peut être modifiée par incréments de 5 à 10 nm et si le pic en question se déplace avec la longueur d’onde d’excitation et diminue en intensité, alors ce pic est dû à la diffusion Raman. Vous pouvez également vérifier si le pic se trouve dans le spectre de solvant vierge. Si c’est le cas, il y a une chance que ce soit un pic Raman. Si le pic de fluorescence est trop proche ou se chevauche avec la diffusion Raman ou Rayleigh, les largeurs de bande et / ou la longueur d’onde d’excitation peuvent être ajustées pour décaler la diffusion hors du pic de fluorescence. Ces effets sont les plus importants pour les très faibles concentrations de fluorophores et en particulier les solutions hautement diffusantes, comme les protéines, les microsphères, les nanoparticules, ainsi que les solides.
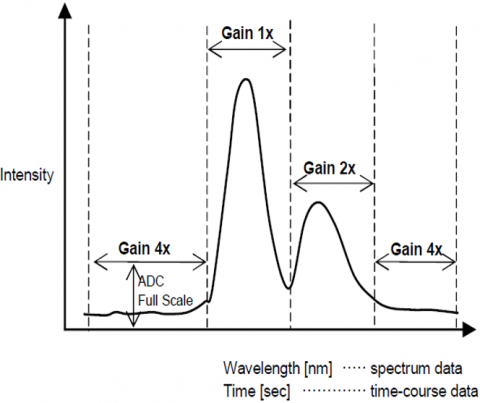
Plage dynamique
La fonction de contrôle automatique du gain ajuste automatiquement le gain d’un signal provenant du détecteur en fonction de l’intensité de fluorescence. Cela optimise le rapport signal / bruit sur toute la plage balayée pour les mesures spectrales ou temporelles, de sorte que les pics d’intensités différentes sont automatiquement ajustés pour améliorer le S / N et assurer la précision des résultats.
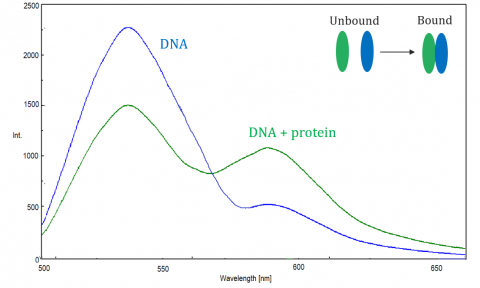
Système de Contrôle Automatique de la sensibilité (SCS)
Le Système de contrôle Automatique de la sensibilité (SCS) élargit la plage dynamique du signal de fluorescence détecté en ajustant automatiquement la tension du détecteur en fonction de l’intensité de fluorescence. Cela permet des mesures de longueurs d’onde fixes ou d’analyses quantitatives de concentrations sub-picomolaires à micromolaires sans changer manuellement l’instrument.

Figure 5. Courbe d’étalonnage des solutions de fluorescéine de 5 * 10-13 à 1,5 · 10-6 M à l’aide de la fonction auto-SCS.
Applications de la spectroscopie de fluorescence
Anisotropie
L’anisotropie de fluorescence est observée lorsqu’un fluorophore émet une lumière d’intensités différentes selon les axes de polarisation et est décrite par l’équation suivante
r = Ivv-GIvh/Ivv +2GIvh
où est l’intensité d’émission parallèle au plan d’excitation et est l’intensité d’émission perpendiculaire au plan d’excitation. G est appelé facteur G ou facteur de réseau d’instruments et tient compte de la dépendance de polarisation du monochromateur d’émission.
Tous les fluorophores ont des moments de transition qui se produisent le long de directions spécifiques le long de l’axe moléculaire. Lorsqu’ils sont exposés à une lumière polarisée, les fluorophores orientés aléatoirement dont les moments de transition d’absorption sont orientés autour de l’angle de la lumière incidente seront excités et cette population d’états excités est partiellement orientée. Lorsqu’une molécule revient d’un état excité à son état fondamental, la charge électronique est redistribuée et le changement d’orientation des moments dipolaires affecte les polarisations d’excitation et d’émission. Par exemple, lorsque la fluorescence est émise avant qu’une molécule ne tourne, la lumière de fluorescence sera fortement polarisée vers la direction de la polarisation de la lumière d’excitation. Si la lumière est émise après la rotation de la molécule dans une direction complètement aléatoire, la fluorescence ne sera plus polarisée.
Lors de la mesure de l’anisotropie de fluorescence, les facteurs suivants affecteront le mouvement moléculaire: (1) la taille moléculaire, (2) la viscosité de l’environnement de la molécule et (3) la force et les degrés de liberté d’une molécule liée. Les mesures d’anisotropie déterminent le déplacement angulaire moyen d’un fluorophore qui se produit entre l’absorption et l’émission d’un photon. Le déplacement angulaire dépend de la vitesse et de l’étendue de la diffusion rotationnelle pendant la durée de vie de l’état excité. Lorsqu’un fluorophore est libre et qu’on le laisse tourner librement avant de réémettre un photon, la vitesse de diffusion est généralement plus rapide que la vitesse de l’émission et l’anisotropie est à peu près égale à zéro. La diffusion rotationnelle change la direction du moment de transition qui dépolarise l’émission. Plus le fluorophore est restreint, plus la valeur d’anisotropie sera grande car la diminution de la flexibilité diminuera la vitesse globale de rotation.
FRET
Le transfert d’énergie par résonance de fluorescence (FRET) est un mécanisme régissant le transfert d’énergie entre deux molécules voisines. Un donneur, initialement dans son état excité, peut transférer de l’énergie à une molécule acceptrice par résonance électronique non radiative.
La FRET est surveillée par le spectrofluoromètre, qui mesure la fluorescence / extinction de l’accepteur ou du donneur excité. L’efficacité de la FRETTE dépend des facteurs suivants: la distance entre le donneur et l’accepteur, le chevauchement spectral entre le donneur et l’accepteur et l’alignement de leurs moments dipolaires. L’efficacité est inversement proportionnelle à la sixième puissance de la distance entre le donneur et l’accepteur, ce qui rend la technique extrêmement sensible aux petits changements de distance. Lorsque la zone de chevauchement du spectre de fluorescence du donneur et du spectre d’absorption de l’accepteur est plus grande, l’efficacité de la FRETTE est plus élevée. L’efficacité de la FRETTE est également maximale lorsque les deux moments dipolaires sont parallèles ou anti-parallèles l’un à l’autre, et aucun transfert d’énergie ne se produit lorsque les moments dipolaires sont perpendiculaires l’un à l’autre. Typiquement, lorsque la distance entre le donneur et l’accepteur est comprise entre 1 et 10 nm, une FRETTE se produit.
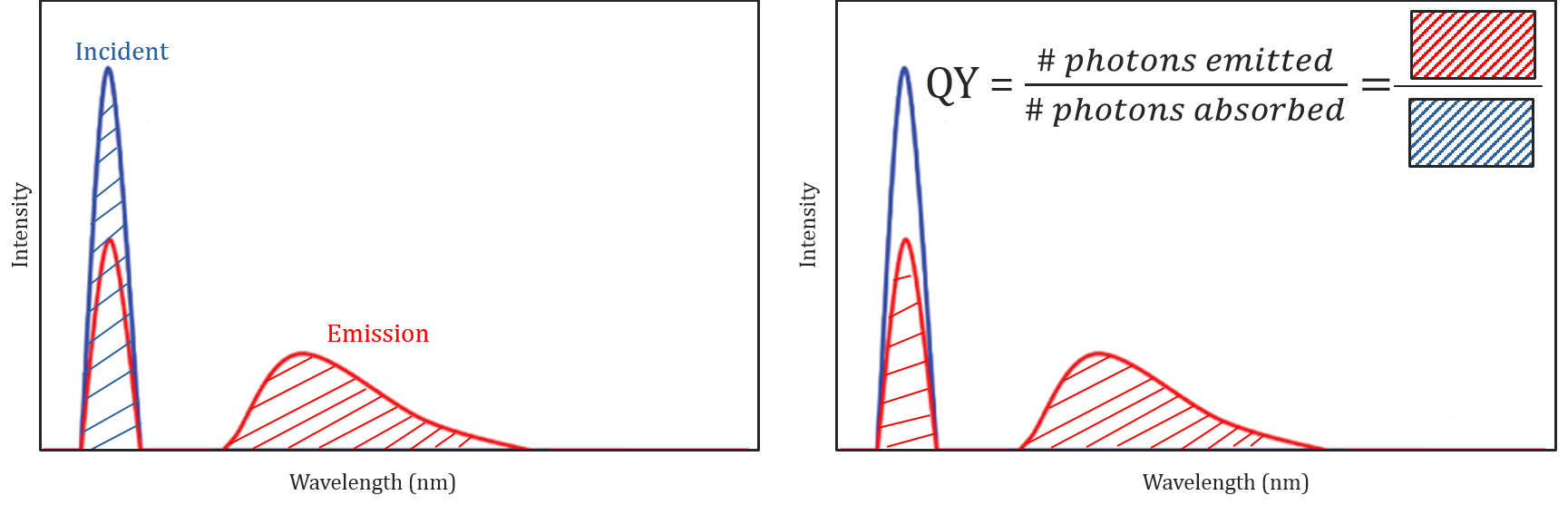
Rendement quantique et Correction spectrale
Différentes conditions moléculaires et environnementales influent non seulement sur la fluorescence ou non d’une molécule, mais peuvent également déterminer l’intensité ou le rendement quantique du rayonnement de fluorescence émis. L’efficacité d’une molécule à la fluorescence est décrite par son rendement quantique et est définie comme le rapport entre le nombre de photons absorbés et le nombre de photons émis par l’échantillon.
Dans certains cas, il est nécessaire de déterminer une mesure spectrale précise. Ceci est fait en utilisant des références à des matériaux calibrés connus. Les sources calibrées utilisées sont vérifiées pour la sortie spectrale absolue sur un instrument connu et un spectre de référence est fourni pour corriger l’instrument individuel fourni au client. Pour que la Correction spectrale fonctionne efficacement, elle doit être effectuée à chaque combinaison de paramètres et de largeur de bande de l’instrument, de sorte que la Correction spectrale à une largeur de bande spectrale de 5 nm ne peut pas être appliquée à la mesure à l’aide d’un SBW de 10 nm. Ceci s’applique à la position des polariseurs s’ils sont utilisés, ainsi qu’à l’utilisation de filtres d’ordre supérieur. Il est nécessaire d’effectuer la Correction spectrale pour chaque combinaison de largeurs de bande spectrales à utiliser par le client, pour l’inclusion ou l’exclusion de la sélection de filtres d’ordre supérieur, et pour les positions des polariseurs s’ils sont montés. Les longueurs d’onde d’excitation et d’émission de l’échantillon détermineront quelle solution / source de lumière est utilisée pour l’étalonnage.
Proche infrarouge
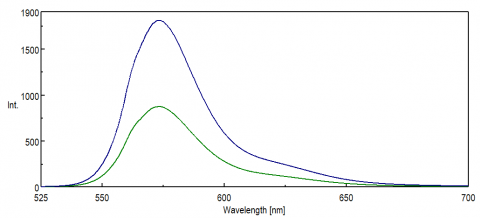
Pour les applications sondant la région NIR du spectre, la réponse spectrale du détecteur PMT est critique pour obtenir des données. Dans l’extrémité rouge de la région visible, dans le NIR, l’efficacité quantique de la PMT diminue de manière significative, ce qui entraîne peu ou pas d’intensité de signal pendant les mesures d’échantillon. Les expériences FRET et les colorants et sondes NIR sont fréquemment surveillés à des longueurs d’onde supérieures à 500 nm et, dans de nombreux cas, ont de petits signaux, même pour une technique aussi sensible que la fluorescence. La figure 8 illustre la différence d’intensité de fluorescence de la rhodamine B en utilisant un PMT standard par rapport à un PMT plus sensible aux longueurs d’onde à l’extrémité rouge du spectre.
Technique complémentaire:
Spectroscopie de dichroïsme circulaire
La spectroscopie de dichroïsme circulaire (CD) est une technique analytique essentielle utilisée pour analyser la chiralité des molécules par leur activité optique. Le CD peut être appliqué à une grande variété de structures moléculaires, mais a trouvé la faveur dans la communauté scientifique pour l’élucidation de la structure macromoléculaire, en particulier des protéines et des acides nucléiques.