Spettroscopia a fluorescenza
La spettroscopia a fluorescenza viene utilizzata di routine per studiare i cambiamenti strutturali nei sistemi coniugati, nelle molecole aromatiche e nei composti rigidi e planari a causa di alterazioni di temperatura, pH, forza ionica, solvente e ligandi. Un singolo fluoroforo può generare migliaia di fotoni rilevabili che possono essere ripetutamente eccitati e rilevati, rendendo la spettroscopia a fluorescenza una tecnica altamente sensibile.
La fluorescenza è un tipo di emissione radiativa che si verifica quando una molecola assorbe energia ad una lunghezza d’onda in cui ha un momento di dipolo di transizione. L’energia di eccitazione fornita alla molecola allo stato fondamentale promuove i fotoni in uno stato di singoletto eccitato, dove poi decadono al livello di energia vibrazionale più basso di questo stato di singoletto eccitato. Questa energia si rilassa ulteriormente allo stato fondamentale della molecola, emettendo fotoni nel processo, come mostrato in Figura 1.
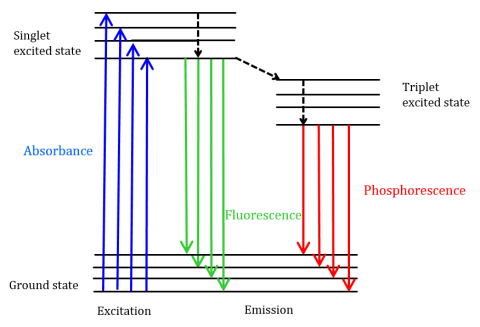
Le molecole fluorescenti possono anche subire tre metodi di rilassamento non radiativo in cui l’energia di eccitazione non viene convertita in fotoni: (1) conversione interna, (2) conversione esterna e (3) incrocio intersistemico. La conversione interna si verifica quando c’è un divario energetico relativamente piccolo tra due stati elettronici e la transizione degli elettroni da uno stato elettronico più alto a uno di energia inferiore. Qui l’energia viene trasferita alle modalità vibrazionali dello stato elettronico. Poiché i processi vibrazionali sono guidati termicamente, l’aumento della temperatura porta a diminuzioni dell’intensità della fluorescenza. Nella conversione esterna, l’energia viene persa attraverso la tempra collisionale con molecole di soluto nell’ambiente del fluoroforo. Intersystem crossing si verifica quando i livelli vibrazionali degli stati eccitati singoletto e tripletta si sovrappongono in energia ed elettroni transizione dal più basso stato eccitato singoletto al primo stato di tripletta eccitato. I fotoni emessi mentre ritornano indietro allo stato fondamentale è noto come fosforescenza (Figura 1). Lo stato della tripletta è più basso nell’energia che lo stato del singoletto in modo dai picchi di fosforescenza sono trovati alle lunghezze d’onda più lunghe che fluorescenza. Poiché anche queste transizioni sono proibite, la fosforescenza presenta una durata maggiore (~10-4 – 102 secondi) rispetto alla fluorescenza (~10-9-10-6 secondi). Le durate più lunghe portano anche alla disattivazione termica tramite tempra dell’ossigeno, movimento del solvente e collisione intermolecolare, quindi la fosforescenza in genere non può essere osservata a temperatura ambiente e i campioni devono quindi essere raffreddati a temperatura di azoto liquido.
Effetti della legge e della concentrazione della birra
Mentre l’assorbimento avviene su una scala temporale inferiore a 10-15 secondi, il processo di rilassamento dallo stato eccitato allo stato fondamentale è molto più lento. Pertanto, la fluorescenza può fornire informazioni sulle interazioni di un fluoroforo con molecole e solventi circostanti, a differenza dell’assorbimento.
L’intensità della fluorescenza è direttamente proporzionale all’intensità della luce di eccitazione
F=2.303 * K * I0 * ebc
dove K è una costante basata sulla geometria dello strumento, I0 è l’intensità della luce di eccitazione, e è l’assorbimento molare del fluoroforo, b è la lunghezza del percorso e c è la concentrazione. Poiché l’intensità della fluorescenza non è rapportata all’intensità della luce incidente come con le misurazioni di assorbimento, la sensibilità della fluorescenza è molto maggiore perché non è limitata dalla capacità degli strumenti di distinguere tra l’intensità incidente e quella rilevata. Di conseguenza, sono necessarie concentrazioni più piccole per le misurazioni.
L’equazione di cui sopra è lineare solo quando l’assorbanza del campione è inferiore a 0,05 AU. Se un campione è troppo concentrato, la luce di emissione può essere riassorbita dal fluoroforo, attenuando il segnale di fluorescenza a lunghezze d’onda più corte. La luce di eccitazione può anche non penetrare completamente l’intera larghezza di un campione altamente concentrato, che inoltre condurrà alle intensità diminuite della fluorescenza.
Strumentazione di spettroscopia di fluorescenza
Caratteristiche di uno spettro di fluorescenza
I fluorometri sono composti da un monocromatore di eccitazione ed emissione, consentendo agli utenti di ottenere sia spettri di eccitazione che di emissione. Una misurazione effettuata da un fluorometro è unica per i monocromatori di eccitazione ed emissione del singolo strumento. La fluorescenza è direttamente correlata al flusso luminoso e all’efficienza della misurazione e quindi dipende dal design dello strumento e dai componenti come la sorgente luminosa, l’ottica monocromatica e il tubo fotomoltiplicatore. Ogni sorgente luminosa avrà un’uscita spettrale diversa (sia forma che potenza) che varierà e diminuirà nel corso della vita della sorgente.
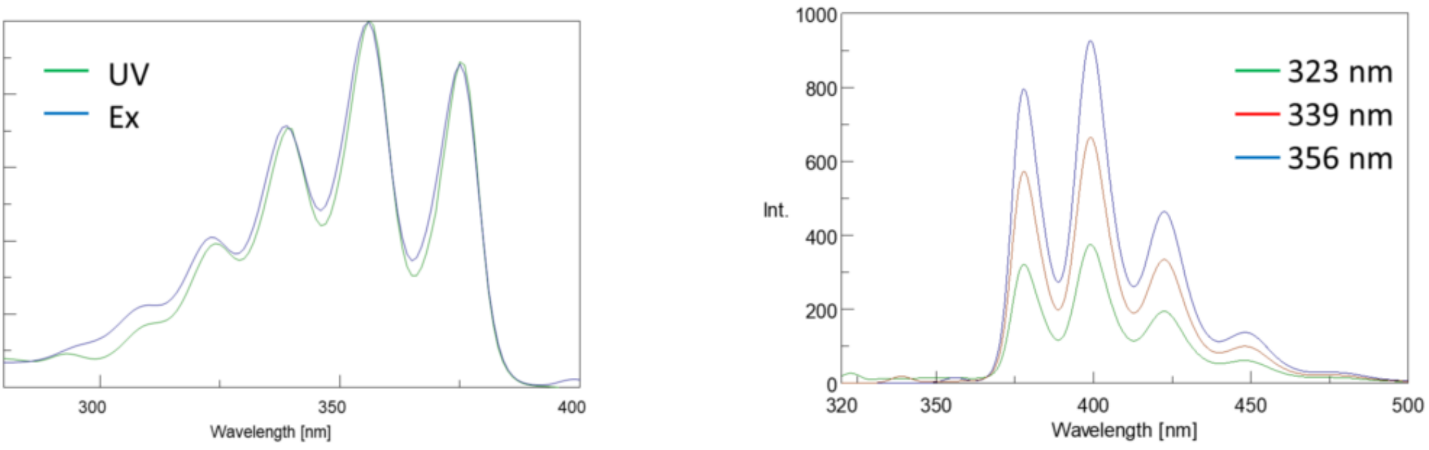
Gli spettri di eccitazione tracciano l’intensità a una lunghezza d’onda di emissione fissa variando le lunghezze d’onda di eccitazione. Poiché la maggior parte degli spettri di emissione sono indipendenti dalla lunghezza d’onda di eccitazione, gli spettri di eccitazione sono spesso duplicati dello spettro di assorbimento del fluoroforo.
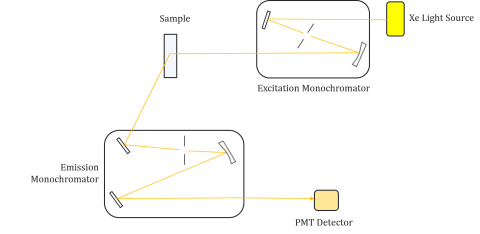
Viceversa, uno spettro di emissione traccia l’intensità a una lunghezza d’onda di eccitazione fissa durante la scansione attraverso lunghezze d’onda di emissione variabili. Queste scansioni di emissione forniscono informazioni sulla struttura molecolare del fluoroforo e sull’ambiente locale che lo circonda. Poiché l’emissione di fluorescenza si verifica sempre dallo stato eccitato più basso allo stato fondamentale, la forma dello spettro di emissione è indipendente dalla lunghezza d’onda di eccitazione. Più energia è necessaria anche per eccitare una molecola da terra allo stato eccitato, con conseguente picchi di emissione a lunghezze d’onda più lunghe (cioè energie più piccole) rispetto alle loro corrispondenti lunghezze d’onda di eccitazione. Questa differenza di energia tra le lunghezze d’onda di eccitazione e di emissione è nota come spostamento di Stokes.
Inoltre, gli spettri di assorbimento ed emissione sono spesso immagini speculari l’una dell’altra a causa dell’uguale distribuzione tra i livelli di energia vibrazionale degli stati eccitati e di terra (Figura 3). Il principio di Franck-Condon spiega che, poiché i nuclei sono relativamente grandi e la transizione elettronica coinvolta nell’emissione e nell’assorbimento avviene su tempi così rapidi, non c’è tempo per i nuclei di muoversi e i livelli di energia vibrazionale e quindi rimangono approssimativamente gli stessi per tutta la transizione elettronica.
Larghezza di banda spettrale
Poiché l’intensità della fluorescenza è proporzionale all’intensità della luce di ingresso, la quantità di luce passata attraverso il monocromatore influenzerà notevolmente l’intensità. La somma delle larghezze di banda di eccitazione ed emissione dovrebbe essere circa la larghezza di banda spettrale (SBW) del picco monitorato in modo che tutti i picchi siano ben risolti. Fino a quando questa regola empirica è seguita, le larghezze di banda possono essere aperte per aumentare la quantità di luce throughput per i campioni con bassa fluorescenza. Il SBW può anche essere influenzato dallo spostamento di Stokes del fluoroforo. Turni di Stokes più stretti possono limitare la gamma di SBW accettabili che possono essere utilizzati.
Artefatti di fluorescenza
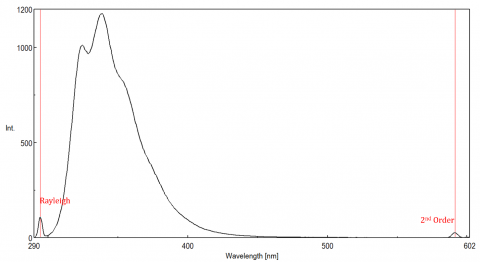
La luce diffusa può dare origine a artefatti, distorcendo lo spettro di fluorescenza. I tre tipi più comuni di dispersione visto in fluorescenza sono Rayleigh, 2 ° ordine, e Raman scatter (Figura 3). Rayleigh scattering è la luce di eccitazione diffusa e quindi picchi alla lunghezza d’onda di eccitazione. la dispersione di 2 ° ordine è una dispersione di ordine superiore osservata al doppio della lunghezza d’onda di eccitazione. Raman scattering è dispersione anelastica a causa di solventi e picchi ad un’energia fissa dalla lunghezza d’onda di eccitazione. Per differenziare lo scattering Raman da un picco di fluorescenza, la lunghezza d’onda di eccitazione può essere variata in incrementi da 5 a 10 nm e se il picco in questione si sposta con la lunghezza d’onda di eccitazione e diminuisce di intensità, allora quel picco è dovuto allo scatter Raman. È inoltre possibile controllare per vedere se il picco è nello spettro solvente vuoto. Se lo è, c’è una possibilità che sia un picco Raman. Se il picco di fluorescenza è troppo vicino o si sovrappone allo scatter Raman o Rayleigh, le larghezze di banda e/o la lunghezza d’onda di eccitazione possono essere regolate per spostare lo scatter dal picco di fluorescenza. Questi effetti sono più importanti per concentrazioni molto basse di fluoroforo e soprattutto soluzioni altamente scattering, come proteine, microsfere, nanoparticelle, così come i solidi.
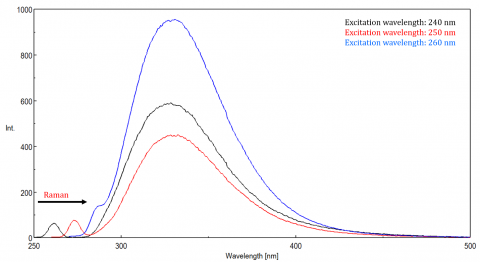
Gamma dinamica
La funzione di controllo automatico del guadagno regola automaticamente il guadagno di un segnale dal rivelatore in base all’intensità della fluorescenza. Ciò ottimizza il segnale-rumore in tutta la gamma scansionata per le misure spettrali o del corso di tempo in modo che i picchi con intensità differenti siano regolati automaticamente per migliorare il S/N ed assicurare l’accuratezza del risultato.
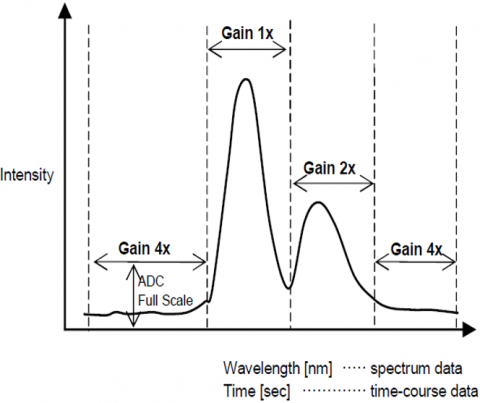
Sistema di controllo automatico della sensibilità(SCS)
Il sistema di controllo automatico della sensibilità (SCS) espande la gamma dinamica del segnale di fluorescenza rilevato regolando automaticamente la tensione del rivelatore in base all’intensità della fluorescenza. Ciò consente misure di lunghezza d’onda fissa o analisi quantitative di concentrazioni sub-picomolari a micromolari senza modificare manualmente lo strumento.

Figura 5. Curva di calibrazione delle soluzioni di fluoresceina da 5 * 10-13 a 1,5 * 10-6 M utilizzando la funzione auto-SCS.
Applicazioni della Spettroscopia di Fluorescenza
Anisotropia
anisotropia di Fluorescenza è osservato quando un fluoroforo emette luce di diversa intensità a seconda delle assi di polarizzazione ed è descritto dalla seguente equazione
r=Ivv-GIvh/Ivv+2GIvh
dove è l’intensità di emissione parallela all’eccitazione piano e l’intensità di emissione perpendicolare all’eccitazione piano. G è chiamato fattore G o fattore di griglia dello strumento e rappresenta la dipendenza di polarizzazione del monocromatore di emissione.
Tutti i fluorofori hanno momenti di transizione che si verificano lungo direzioni specifiche lungo l’asse molecolare. Quando esposti alla luce polarizzata, i fluorofori orientati casualmente che hanno i loro momenti di transizione di assorbimento orientati attorno all’angolo della luce incidente saranno eccitati e questa popolazione di stato eccitato è parzialmente orientata. Quando una molecola ritorna da uno stato eccitato al suo stato fondamentale, la carica dell’elettrone viene ridistribuita e il cambiamento nell’orientamento dei momenti di dipolo influisce sulle polarizzazioni di eccitazione ed emissione. Ad esempio, quando la fluorescenza viene emessa prima che una molecola ruoti, la luce a fluorescenza sarà fortemente polarizzata verso la direzione della polarizzazione della luce di eccitazione. Se la luce viene emessa dopo la rotazione della molecola in una direzione completamente casuale, la fluorescenza non sarà più polarizzata.
Quando si misura l’anisotropia della fluorescenza, i seguenti fattori influenzeranno il movimento molecolare: (1) dimensione molecolare, (2) viscosità dell’ambiente della molecola e (3) forza e gradi di libertà di una molecola legata. Le misurazioni dell’anisotropia determinano lo spostamento angolare medio di un fluoroforo che si verifica tra l’assorbimento e l’emissione di un fotone. Lo spostamento angolare dipende dalla velocità e dall’estensione della diffusione rotazionale durante la vita dello stato eccitato. Quando un fluoroforo non ha restrizioni e può ruotare liberamente prima di riemettere un fotone, la velocità di diffusione è generalmente più veloce della velocità dell’emissione e l’anisotropia è approssimativamente uguale a zero. La diffusione rotazionale cambia la direzione del momento di transizione che depolarizza l’emissione. Più limitato è il fluoroforo, maggiore sarà il valore di anisotropia poiché la diminuzione della flessibilità diminuirà la velocità complessiva di rotazione.
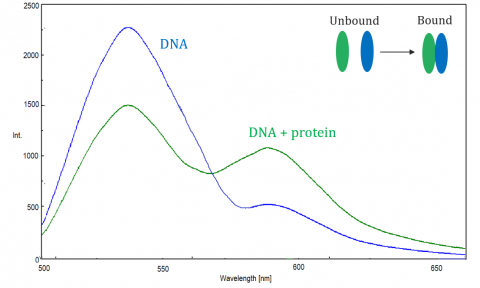
FRET
Il trasferimento di energia a risonanza di fluorescenza (FRET) è un meccanismo che governa il trasferimento di energia tra due molecole vicine. Un donatore, inizialmente nel suo stato eccitato, può trasferire energia a una molecola accettore attraverso risonanza elettronica non radiativa.
Il TASTO è monitorato dallo spettrofluorometro, che misura la fluorescenza / tempra dell’accettore o del donatore eccitato. L’efficienza del TASTO dipende dai seguenti fattori: la distanza tra il donatore e l’accettore, la sovrapposizione spettrale tra il donatore e l’accettore e l’allineamento dei loro momenti di dipolo. L’efficienza è inversamente proporzionale alla sesta potenza della distanza tra donatore e accettore, rendendo la tecnica estremamente sensibile a piccoli cambiamenti di distanza. Quando l’area di sovrapposizione dello spettro di fluorescenza del donatore e dello spettro di assorbimento dell’accettore è più grande, l’efficienza del TASTO è più alta. L’efficienza del TASTO è anche al massimo quando i due momenti di dipolo sono paralleli o anti-paralleli tra loro, e nessun trasferimento di energia si verifica quando i momenti di dipolo sono perpendicolari l’uno all’altro. Tipicamente quando la distanza tra il donatore e l’accettore è betwen1 e 10 nm, FRET si verifica.
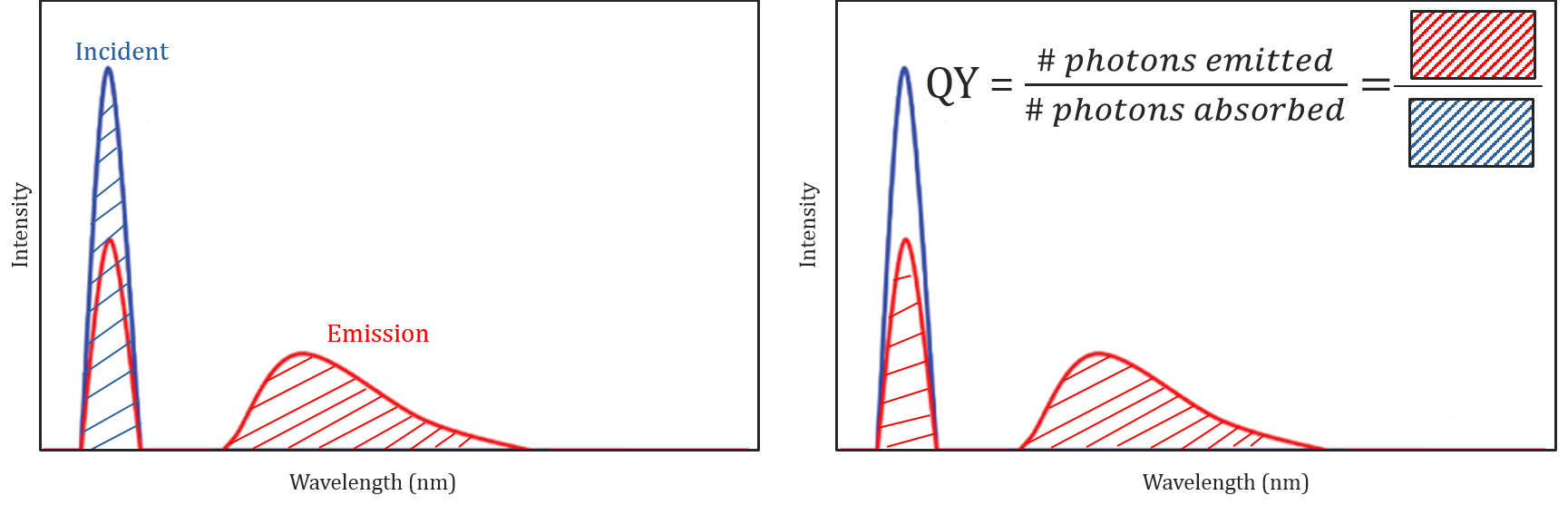
Resa quantistica e correzione spettrale
Diverse condizioni molecolari e ambientali non solo influenzano se una molecola fluoresce o meno, ma possono anche determinare l’intensità o la resa quantistica della radiazione di fluorescenza emessa. L’efficienza di una molecola a fluorescenza è descritta dalla sua resa quantistica ed è definita come il rapporto tra il numero di fotoni assorbiti e il numero di fotoni emessi dal campione.
In alcuni casi è necessario determinare una misurazione spettrale accurata. Questo è fatto usando riferimenti a materiali calibrati noti. Le sorgenti calibrate utilizzate vengono controllate per l’uscita spettrale assoluta su uno strumento noto e viene fornito uno spettro di riferimento per correggere il singolo strumento fornito al cliente. Affinché la correzione spettrale funzioni in modo efficace, deve essere eseguita su ogni parametro dello strumento e sulle combinazioni di larghezza di banda in modo che la correzione spettrale a una larghezza di banda spettrale di 5 nm non possa essere applicata alla misurazione utilizzando un SBW di 10 nm. Questo vale per la posizione dei polarizzatori se vengono utilizzati, così come l’uso di filtri di ordine superiore. È necessario eseguire la Correzione spettrale per ogni combinazione di larghezze di banda spettrali da utilizzare dal cliente, per l’inclusione o l’esclusione della selezione del filtro di ordine superiore e per le posizioni dei polarizzatori se montati. Le lunghezze d’onda di eccitazione e di emissione del campione determineranno quale soluzione/sorgente luminosa viene utilizzata per la calibrazione.
Vicino infrarosso
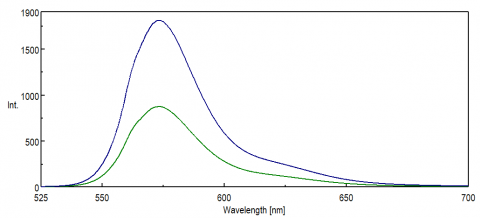
Per applicazioni che sondano la regione NIR dello spettro, la risposta spettrale del rivelatore PMT è fondamentale per ottenere dati. Nell’estremità rossa della regione visibile, nel NIR, l’efficienza quantistica del PMT diminuisce significativamente, con conseguente poca o nessuna intensità del segnale durante le misurazioni del campione. Esperimenti FRET e coloranti NIR e sonde sono spesso monitorati a lunghezze d’onda superiori a 500 nm e in molti casi hanno piccoli segnali, anche per una tecnica così sensibile come la fluorescenza. La figura 8 illustra la differenza di intensità di fluorescenza della rodamina B utilizzando un PMT standard rispetto a un PMT più sensibile alle lunghezze d’onda all’estremità rossa dello spettro.
Tecnica complementare:
Spettroscopia dicroica circolare
La spettroscopia dicroica circolare (CD) è una tecnica analitica essenziale utilizzata per analizzare la chiralità nelle molecole attraverso la loro attività ottica. Il CD può applicarsi ad un’ampia varietà di strutture molecolari ma ha trovato il favore nella comunità scientifica per la delucidazione della struttura macromolecolare, particolarmente proteine ed acidi nucleici.