Differential expression analysisとは、正規化された読み取りカウントデータを取得し、統計解析を実行して、実験グループ間の発現レベルの定量的変化を発見 例えば、我々は、与えられた遺伝子について、観察された読み取り数の差が有意であるかどうか、すなわち、それが自然なランダムな変動のためだけに予想されるものよりも大きいかどうかを決定するために統計的検定を使用する。
微分発現解析の方法
微分発現解析には、負の二項分布(NB)に基づくedgeRとDESeq、または負の二項モデルに基づくベイズアプローチであるbaySeqとEBSeqなどの異な 解析方法を選択する際には、実験計画を考慮することが重要です。 差分式ツールの中にはペア単位の比較しか実行できないものもありますが、edgeR、limma-voom、DESeq、maSigProなどの他のツールは多重比較を実行できます。
以下の図11では、Expression Atlasのデータを生成するために使用されるRNA-seq処理パイプラインの概要を説明します。
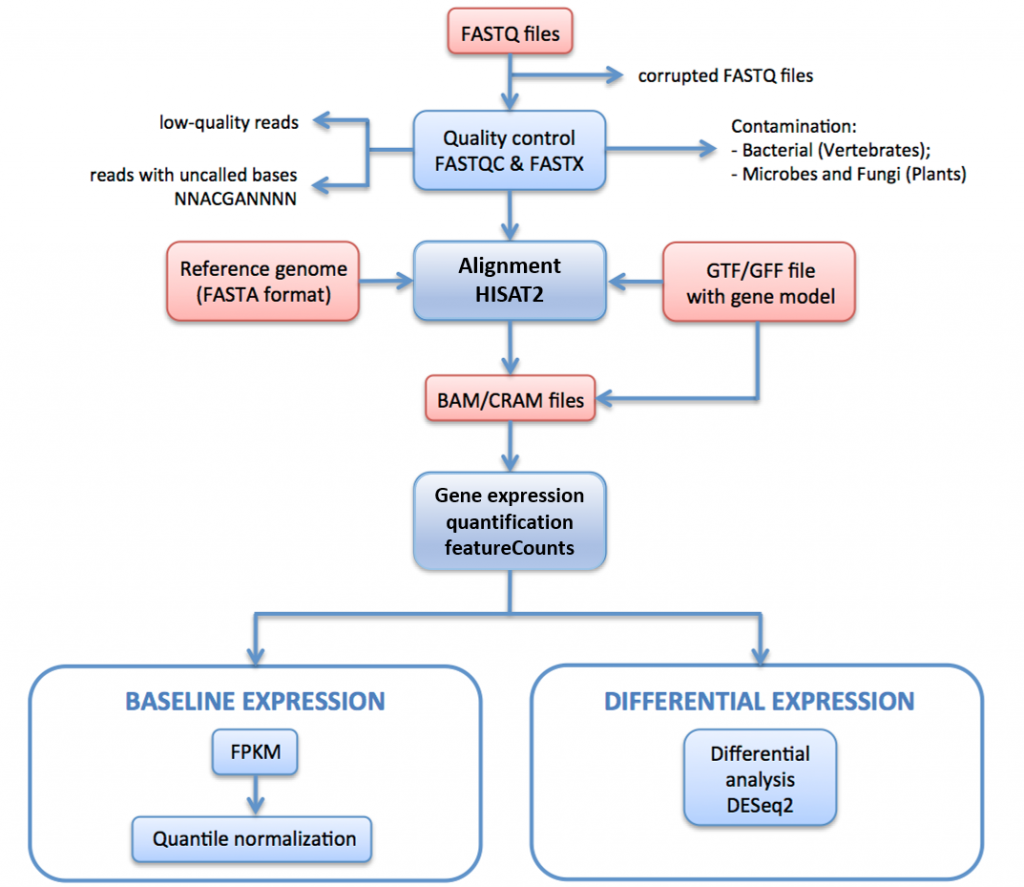
このパイプラインでは、raw reads(FASTQファイル)は品質評価とフィルタリングを受けます。 品質フィルタされた読み取りは、HISAT2を介して参照ゲノムに整列される。 マップされた読み取りは、HTSeqを介して遺伝子上に要約され、集約される。 ベースライン式の場合、FPKMは、IRAPによって生の計数から計算される。 これらは、技術的複製の各セットについて平均化され、次いで、limmaを使用して生物学的複製の各セット内で正規化された分位点である。