różnicowa analiza ekspresji oznacza pobranie znormalizowanych danych liczby odczytów i przeprowadzenie analizy statystycznej w celu wykrycia ilościowych zmian poziomów ekspresji między grupami doświadczalnymi. Na przykład, używamy testów statystycznych, aby zdecydować, czy dla danego genu zaobserwowana różnica w liczbach odczytu jest znacząca, to znaczy, czy jest większa niż to, czego można by się spodziewać tylko z powodu naturalnej zmienności losowej.
metody analizy ekspresji różniczkowej
istnieją różne metody analizy ekspresji różniczkowej, takie jak edgeR i DESeq oparte na ujemnych rozkładach dwumianowych (NB) lub baySeq i EBSeq, które są podejściem Bayesowskim opartym na ujemnym modelu dwumianowym. Ważne jest, aby wziąć pod uwagę projekt eksperymentalny przy wyborze metody analizy. Podczas gdy niektóre narzędzia do wyrażania różnicowego mogą wykonywać tylko porównywanie par, inne, takie jak edgeR, limma-voom, DESeq i maSigPro, mogą wykonywać wiele porównań.
na rysunku 11 poniżej przedstawiamy proces przetwarzania RNA-seq używany do generowania danych dla Atlasu ekspresji.
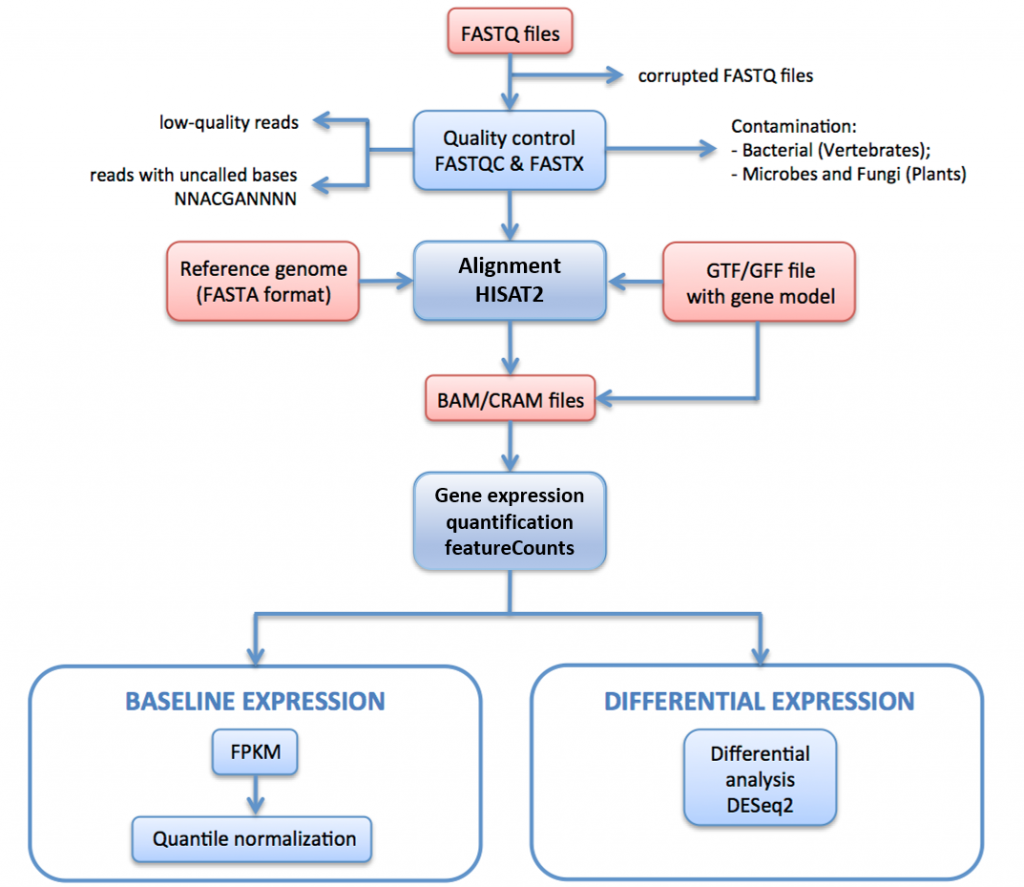
w tym potoku surowe odczyty (pliki FASTQ) są poddawane ocenie jakości i filtrowaniu. Przefiltrowane jakościowo odczyty są dopasowane do genomu referencyjnego poprzez HISAT2. Zmapowane odczyty są sumowane i agregowane w genach za pomocą HTSeq. Dla wyrażenia bazowego, Fpkm są obliczane z surowych zliczeń przez iRAP. Są one uśrednione dla każdego zestawu technicznych replikatów, a następnie kwantyl znormalizowany w każdym zestawie biologicznych replikatów za pomocą limma.