differentiële expressieanalyse betekent dat de genormaliseerde leesgegevens worden gebruikt en statistische analyses worden uitgevoerd om kwantitatieve veranderingen in expressieniveaus tussen experimentele groepen te ontdekken. Bijvoorbeeld, we gebruiken statistische testen om te beslissen of, voor een bepaald gen, een waargenomen verschil in het lezen telt significant is, dat wil zeggen, of het groter is dan wat zou worden verwacht gewoon als gevolg van natuurlijke willekeurige variatie.
methoden voor differentiële expressieanalyse
er zijn verschillende methoden voor differentiële expressieanalyse, zoals edgeR en DESeq gebaseerd op negatieve binomiale (NB) distributies of baySeq en EBSeq die Bayesiaanse benaderingen zijn gebaseerd op een negatief binomiaal model. Het is belangrijk om het experimentele ontwerp te overwegen bij het kiezen van een analysemethode. Terwijl sommige van de differentiële expressiehulpmiddelen alleen paarsgewijze vergelijking kunnen uitvoeren, kunnen anderen zoals edgeR, limma-voom, deseq en maSigPro meerdere vergelijkingen uitvoeren.
in Figuur 11, hieronder, schetsen we de RNA-seq verwerkingspijplijn die wordt gebruikt om gegevens voor Expressieatlas te genereren.
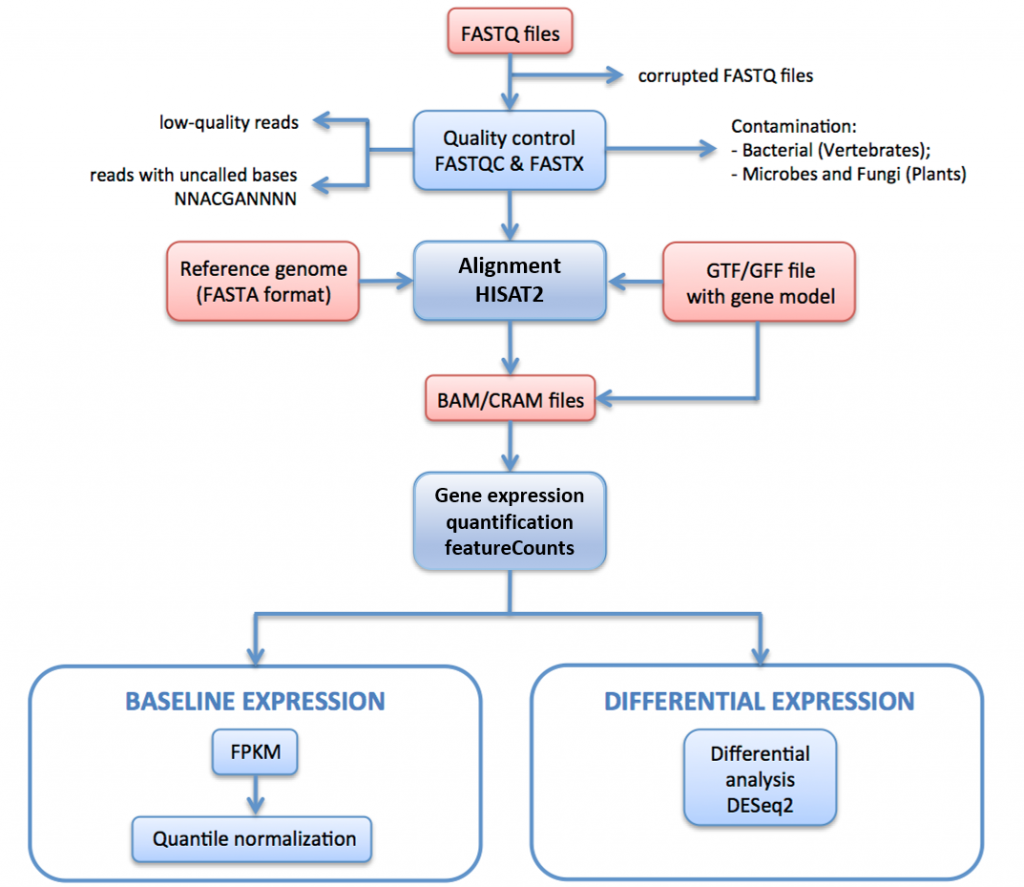
in deze pijplijn raw leest (fastq bestanden) ondergaan kwaliteitsbeoordeling en filtering. De kwaliteit-gefiltreerde leest worden afgestemd op het verwijzingsgenoom via HISAT2. De in kaart gebrachte leest worden samengevat en samengevoegd over genen via HTSeq. Voor baseline expressie worden de Fpkm ‘ s berekend op basis van de ruwe tellingen door iRAP. Deze worden gemiddeld voor elke reeks technische replicaten, en vervolgens genormaliseerd quantiel binnen elke reeks biologische replicaten gebruikend limma.