Differentialuttrycksanalys innebär att man tar normaliserade läsantalsdata och utför statistisk analys för att upptäcka kvantitativa förändringar i uttrycksnivåer mellan experimentella grupper. Till exempel använder vi statistisk testning för att avgöra om en observerad skillnad i läsantal för en given gen är signifikant, det vill säga om den är större än vad som förväntas bara på grund av naturlig slumpmässig variation.
metoder för differentialuttrycksanalys
det finns olika metoder för differentialuttrycksanalys såsom edgeR och deseq baserat på negativa binomiala (NB) fördelningar eller baySeq och EBSeq som är Bayesianska tillvägagångssätt baserade på en negativ binomialmodell. Det är viktigt att överväga den experimentella designen när man väljer en analysmetod. Medan vissa av differentialuttrycksverktygen bara kan utföra parvis jämförelse, kan andra som edgeR, limma-voom, deseq och maSigPro utföra flera jämförelser.
i Figur 11 nedan beskriver vi RNA-seq-bearbetningsrörledningen som används för att generera data för Uttrycksatlas.
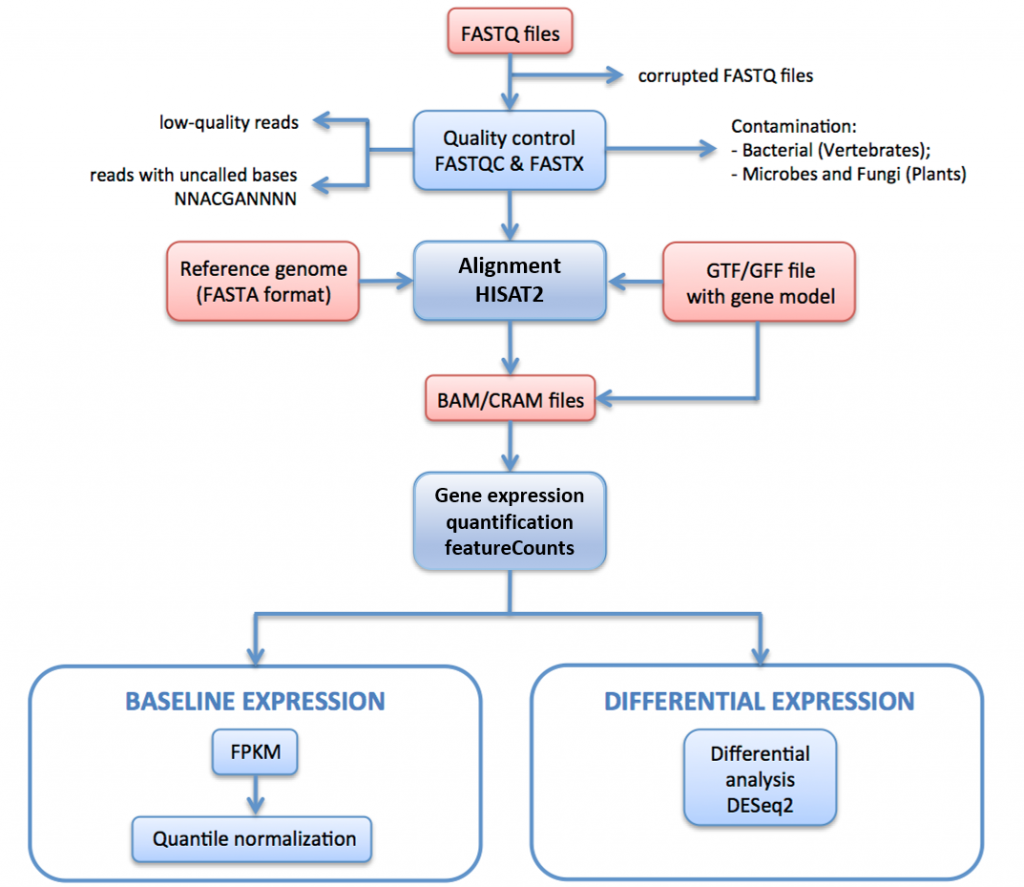
i denna pipeline raw läser (FASTQ filer) genomgår kvalitetsbedömning och filtrering. De kvalitetsfiltrerade läsningarna är anpassade till referensgenomet via HISAT2. De mappade läsningarna sammanfattas och aggregeras över gener via HTSeq. För baslinjeuttryck beräknas FPKMs från raw-räkningarna med iRAP. Dessa medelvärde för varje uppsättning av tekniska replikat, och sedan kvantil normaliseras inom varje uppsättning av biologiska replikat med hjälp av limma.