El análisis diferencial de expresión significa tomar los datos normalizados de recuento de lecturas y realizar análisis estadísticos para descubrir cambios cuantitativos en los niveles de expresión entre los grupos experimentales. Por ejemplo, utilizamos pruebas estadísticas para decidir si, para un gen dado, una diferencia observada en el recuento de lecturas es significativa, es decir, si es mayor de lo que se esperaría solo debido a la variación aleatoria natural.
Métodos para el análisis de expresiones diferenciales
Existen diferentes métodos para el análisis de expresiones diferenciales, como edgeR y DESeq basados en distribuciones binomiales negativas (NB) o baySeq y EBSeq, que son enfoques bayesianos basados en un modelo binomial negativo. Es importante tener en cuenta el diseño experimental al elegir un método de análisis. Mientras que algunas de las herramientas de expresión diferencial solo pueden realizar comparaciones de pares, otras como edgeR, limma-voom, DESeq y maSigPro pueden realizar comparaciones múltiples.
En la Figura 11, a continuación, describimos la tubería de procesamiento ARN-seq utilizada para generar datos para Expression Atlas.
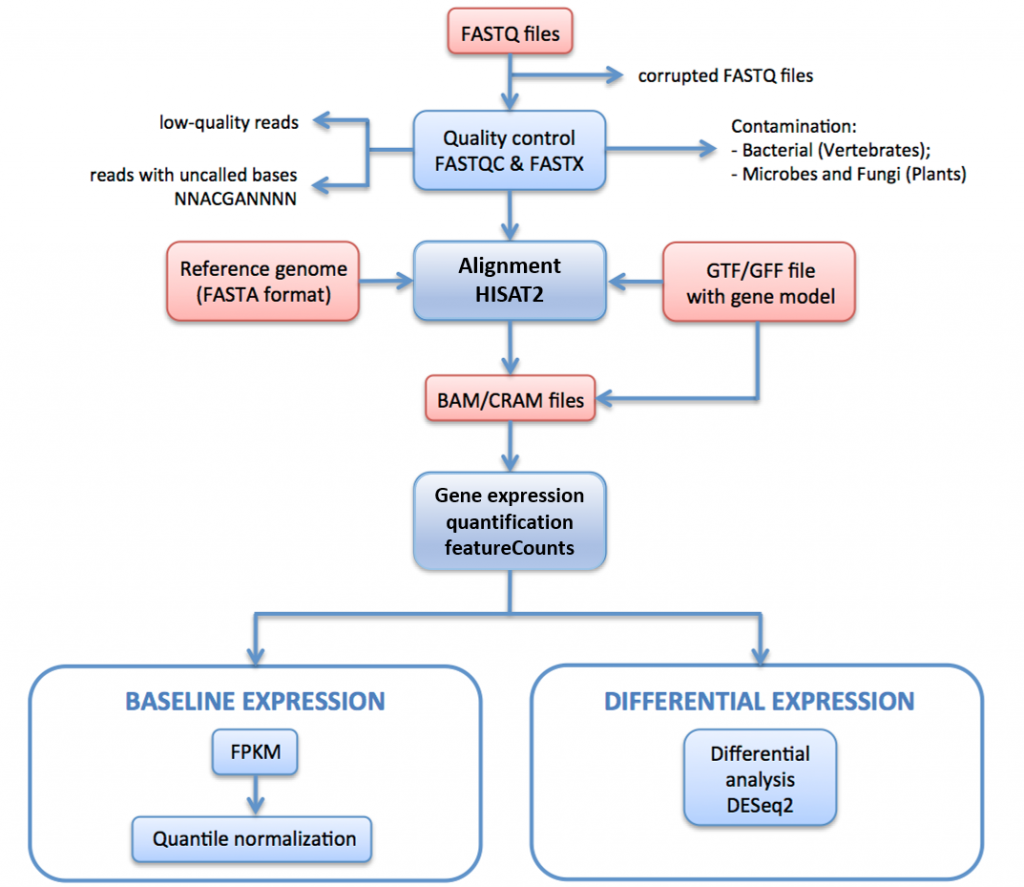
En esta canalización, las lecturas raw (archivos FASTQ) se someten a una evaluación de calidad y filtrado. Las lecturas filtradas por calidad se alinean con el genoma de referencia a través de HISAT2. Las lecturas mapeadas se resumen y agregan sobre genes a través de HTSeq. Para la expresión de línea de base, los FPKM se calculan a partir de los recuentos sin procesar de iRAP. Estos se promedian para cada conjunto de réplicas técnicas, y luego se normalizan cuantiles dentro de cada conjunto de réplicas biológicas utilizando limma.