L’analyse d’expression différentielle consiste à prendre les données de comptage de lecture normalisées et à effectuer une analyse statistique pour découvrir les changements quantitatifs des niveaux d’expression entre les groupes expérimentaux. Par exemple, nous utilisons des tests statistiques pour décider si, pour un gène donné, une différence observée dans le nombre de lectures est significative, c’est-à-dire si elle est supérieure à ce à quoi on s’attendrait simplement en raison d’une variation aléatoire naturelle.
Méthodes d’analyse d’expression différentielle
Il existe différentes méthodes d’analyse d’expression différentielle telles que edgeR et DESeq basées sur des distributions binomiales négatives (NB) ou baySeq et EBSeq qui sont des approches bayésiennes basées sur un modèle binomial négatif. Il est important de prendre en compte le plan expérimental lors du choix d’une méthode d’analyse. Alors que certains outils d’expression différentielle ne peuvent effectuer que des comparaisons par paires, d’autres tels que edgeR, limma-voom, DESeq et maSigPro peuvent effectuer plusieurs comparaisons.
Dans la figure 11, ci-dessous, nous décrivons le pipeline de traitement de l’ARN-seq utilisé pour générer des données pour l’Atlas d’expression.
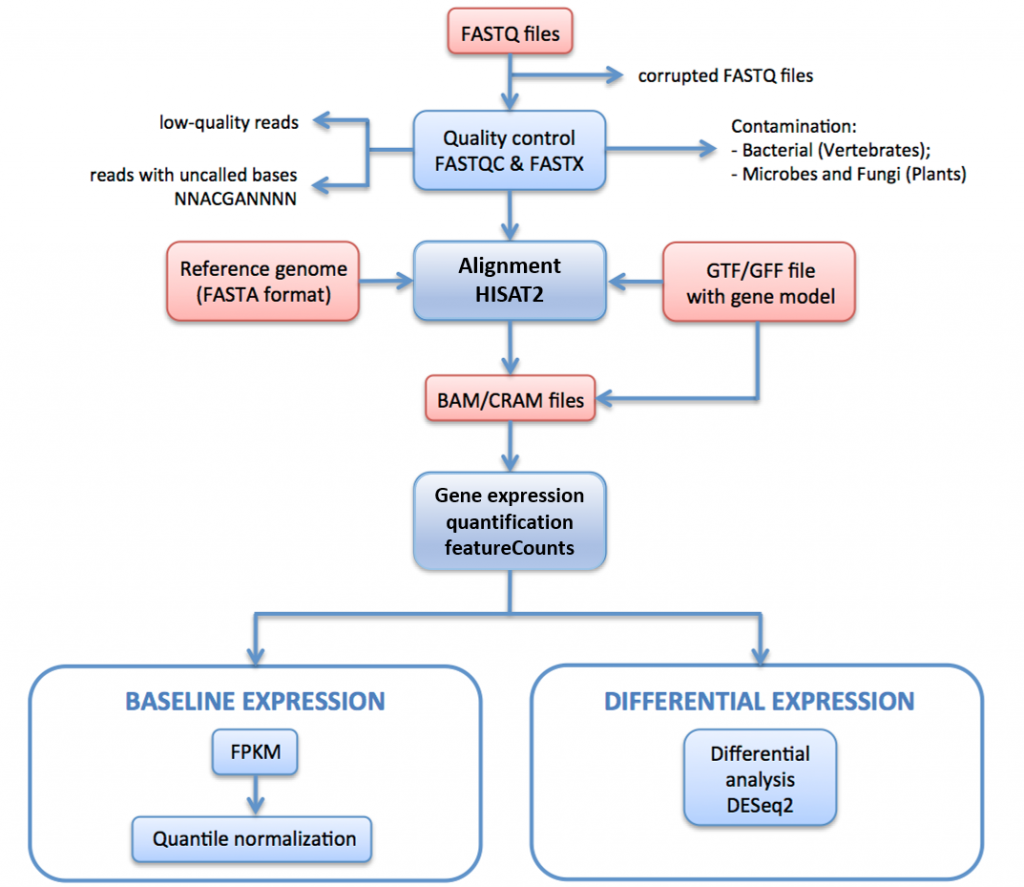
Dans ce pipeline, les lectures brutes (fichiers FASTQ) subissent une évaluation de la qualité et un filtrage. Les lectures filtrées par la qualité sont alignées sur le génome de référence via HISAT2. Les lectures cartographiées sont résumées et agrégées sur les gènes via HTSeq. Pour l’expression de base, les FPKMs sont calculés à partir des comptes bruts par iRAP. Ceux-ci sont moyennés pour chaque ensemble de répliques techniques, puis normalisés en quantile dans chaque ensemble de répliques biologiques à l’aide de limma.